





بیماری Spinal muscular atrophy (SMA) یک بیماری ارثی و یکی از انواع بیماریهای نوروماسکولار همچون دیستروفی میباشد. این بیماری در چهار فرم که در شدت علائم و سن بروز متفاوت هستد طبقه بندی می شود.
SMA نوع 1(SMA I): به این نوع بیماری که شدیدترین و رایج ترین نوع SMA است “وردینگ هافمن” نیز گفته می شود .SMA I معمولا در بدو تولد یا در چند ماه اول پس از آن (6-0 ماه) مشهود است. کودکان با این نوع معمولاً توانایی حرکتی بسیار محدودی دارند. همچنین در تغذیه و بلعیدن ، بالا نگه داشتن سر و تنفس دچار مشکل خواهند شد. SMA I به سرعت پیشرفت می کند ، با ضعیف شدن عضلات منجر به عفونت های مکرر تنفسی و معمولاً مرگ در سن 2 سالگی می شود. نوزادان مبتلا به این فرم بیماری هرگز نمی توانند بنشینند.
SMA نوع 2 (SMA II): علائم معمولاً متوسط و در سنین 7 تا 18 ماهگی ظاهر می شوند. میزان پیشرفت می تواند بسیار متفاوت باشد. این بیماری روی پاهای کودک تأثیر می گذارد. کودکان مبتلا به این فرم هرگز نمی توانند بایستند. عفونت های تنفسی نیز با این نوع SMA شایع است. امید به زندگی می تواند از اوایل کودکی تا بزرگسالی بسته به شدت وضعیت بیمار متفاوت باشد.
SMA نوع 3 (SMA III ): به این نوع SMA آتروفی عضلانی نخاع “کاگلبرگ-والندر” یا آتروفی عضلانی نخاع “نوجوانی” نیز گفته می شود. علائم ابتدا می توانند در طی طیف گسترده ای از سن ، از 18 ماهگی تا اوایل بزرگسالی و به صورت خفیف ظاهر شوند. بیماران مبتلا SMA III می توانند بایستند و راه بروند ، اما ممکن است در برخاستن از حالت نشسته مشکل داشته باشند. همچنین ممکن است ضعف عضلانی خفیف را تجربه کنند و بیشتر در معرض خطر عفونت های تنفسی هستند. بیشتر بیماران به این فرم از بیماری امید به زندگی نزدیک طبیعی دارند.
SMA نوع 4 (SMA IV ): علائم این نوع نادر SMA معمولاً تا دهه دوم یا سوم زندگی بروز نمی کند. بیماران SMA IV می توانند در بزرگسالی راه بروند اما معمولاً ضعف عضلانی پیشرونده و سایر علائم معمولی SMA را تجربه می کنند.( Adult )
علل بیماری SMA
سلولهای عصبی موجود در قاعده مغز و شاخ جلویی نخاع در ایجاد حرکات بدن نقش اصلی دارند. از بین رفتن تدریجی این سلول ها سبب ایجاد بیماری SMA می شود. این بیماری در واقع بعلت عدم تعادل در مرگ برنامه ریزی شده (آپوپتوز) سلولهای شاخ قدامی نخاع ایجاد می شود.
در بیش از 95 درصد موارد ، SMA ناشی از تولید ناکافی پروتئینی به نام پروتئین نورون حرکتی بقا (SMN) است که برای نورون های حرکتی ضروری است. SMN توسط ژن SMN و به میزان کمتری توسط ژن SMN2 که هردو بر روی کروموزم 5 واقع شده اند تولید می شود.
بیماری SMA اکثرا بهدلیل نقص و جهش در ژن SMN 1 ایجاد میشود . تقریبا در 94٪ موارد، این جهش شامل حذف بخشی است که به عنوان اگزون 7 و 8 شناخته می شود. میزان اندک این پروتئین در سلولها، موجب از دست رفتن عملکرد طبیعی در سلولهای عصبی شاخ قدامی نخاع شده و به صورت عصبی-نخاعی پیشرونده میباشد که با گذشت زمان شدت بیماری نیز افزایش مییابد. بروز این بیماری در دختران و پسران یکسان بوده و به صورت اتوزومی مغلوب به ارث میرسد.
ژنی دیگر در نزدیکی سانترومر کروموزوم 5، به نام SMN2 ، پروتئین SMN را نیز تولید می کند. درصد کمی ، حدود 10 تا 15٪ پروتئین ساخته شده از این ژن عملکردی است.
افراد می توانند چندین نسخه از ژن SMN2 داشته باشند. به طور معمول ، تعداد آنها بین صفر تا هشت نسخه متفاوت است. در واقع، هرچه تعداد کپی از ژن SMN2 در شخص بیشتر باشد ، عملکرد پروتئین SMN بیشتر است. در نتیجه ، احتمالاً روند بیماری خفیف تر خواهد بود. داشتن سه یا چند نسخه از ژن SMN2 با تظاهرات بالینی خفیف بیماری همراه است.
نحوه تشخیص بیماری SMA
تشخیص بالینی بیماری SMA سخت بوده و معمولا این بیماری همانطور که در توضیحات هر یک از انواع آن ارائه شد دارای علائم گوناگونی است. اگر کودکی با این مشخصات را میشناسید سعی کنید با والدین وی صحبت کنید تا سریعا به یک پزشک متخصص مراجعه کنند. در صورتی که بیمار دارای ضعف در عضلات است، اگر در معاینه صورت گرفته، رفلکسهای تاندونهای عمقی شخص وجود نداشته باشند باید به وقوع این بیماری شک کرد که در این صورت لازم است تا آنزیمهای عضلات چک و بررسی شوند.
همچنین برای تشخیص SMA لازم است ابتدا نوار عصب و عضله برای بیمار تهیه شود. به دلیل اینکه این بیماری عصبی-عضلانی میباشد تهیه نوار عصب و عضله میتواند نمای خاصی از این بیماری را نشان داده و به عنوان کم هزینهترین راه برای تشخیص بیماری مورد استفاده قرار گیرد. برای تشخیص قطعی بیماری SMA آزمایش ژنتیک لازم است تا در آن به بررسی ژنهایSMA بیمار پرداخته شود. انجام آزمایش نیز با گرفتن خون بیمار صورت میگیرد. سپس با استفاده از تکنیک Multiplex ligation-dependent probe amplification (MLPA) تعداد کپی ژن های SMN 1 و SMN 2 مورد بررسی قرا ر میگیرند.
به دلیل موروثی بودن بیماری لازم است کلیه اعضای خانواده درجه اول و دوم در هنگام ازدواج و بارداری نیز مورد بررسی قرار گیرند تا نقص ژنی در خانواده مورد بررسی قرار گرفته و در نهایت از انتقال بیماری به نسل بعد جلوگیری شود.
بررسی ابتلا به بیماری SMA در دو سطح می تواند مورد بررسی قرار گیرد که عبارتند از:
- تشخیص بیماری با گرفتن خون زوجین آقا و خانم، پدر و مادر بیمار و یا خود بیمار جهت ناقل یا مبتلا بودن
- نمونه گیری در دوران حاملگی که در هفته ۱۰ حاملگی از پرزهای جفتی (CVS) صورت میگیرد
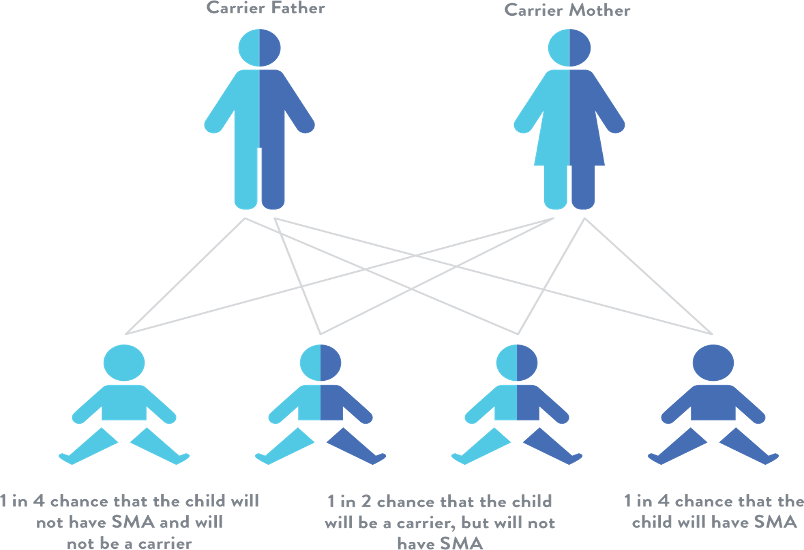
ابتدا باید پدر و مادر جنین آزمایش ژنتیک مربوط به ناقل بودن بیماری را انجام دهند. اگر زوجین ناقل بیماری SMA تشخیص داده شوند ، در این صورت احتمال ابتلا جنین به بیماری ۲۵ درصد خواهد بود. بنابر این انجام آزمایش CVS با صلاح دید پزشک متخصص به جهت اطمینان از سلامت جنین انجام خواهد گرفت. لازم به ذکر است که انجام آزمایش CVS تنها در صورتی انجام می شود که زوجین ناقل بیماری باشند.
درمان بیماری SMA
جایگزینی ژن SMN1 با کمک ژن درمانی ( Gene therapy )
هدف از ژن درمانی در مبتلایان به SMA، بازسازی عملکرد ژنِ معیوب از طریق الحاقِ یک توالی نوکلئوتیدی از پیش ساختهشده (یک ترانس ژن SMN1) به درونِ هسته ی سلول است که از طریق حاملهای ویروسی انجام میشود. حاملهای ویروسی مهمی که هم اکنون در دست پژوهش هستند «scAAV-9» و «scAAV-10» نام دارند. در سال ۲۰۱۹ میلادی، یک درمان مبتنی بر AAV9 مورد پذیرش واقعشد : Onasemnogene abeparvovec
در حال حاضر تنها یکی از پژوهشها به مرحله بالینی رسیدهاست. مطالعات برای انجام ژن درمانی در انستیتو مایولوژی پاریس ودانشگاه آکسفورد در حالِ انجام است. در سال ۲۰۱۸ میلادی، بایوژن هم اعلام کرد که مشغول کار بر روی یک داروی ژن درمانی جهت درمانِ آتروفی عضلانی نخاعی است.
تغییر در پیرایش متناوب SMN2
هدف نهایی در این روش، تغییر دادن نحوه پیرایش متناوب (Alternative Splicing) در ژن SMN2 به نحوی است که منجر به تولید مقادیر بیشتری از پروتئین SMN شود. گاهی به این روش، «تبدیل ژن» هم میگویند، چرا که تلاش میشود تا ژن SMN2 را از لحاظ عملکردی، به ژنِ SMN1 مبدل سازد.
تغییردهندههای پیرایش دگرسانیِ ذیل، به مرحلهٔ آزمایشهای بالینی رسیدهاست:
«براناپلام» (LMI070 و یا NVS-SM1) یک مولکول کوچک ساختگی و آزمایشی است که بهصورت خوراکی تجویز میشود و توسط شرکت دارویی Novartis ساخته شدهاست. تا اکتبر ۲۰۱۷ میلادی، این دارو در فاز ۲ کارآزمایی بالینی در نوزادان مبتلا به نوع ۱ بیماری بودهاست و کارآزماییهای دیگری هم برای سایر مراحل بیماری، در حال طراحی است.
«RG7916» یک مولکول کوچک دارویی است که خوراکی تجویز میشود و با همکاری شرکت دارویی Hoffmann-La Roche و «بنیاد SMA» ساخته شدهاست. تا اکتبر ۲۰۱۶ میلادی، این مولکول در فاز ۲ کارآزمایی بالینی، در تمامی گروههای سنی و تمامی انواع بیماری بوده است.
از میان داروهایی که کارآزمایی هایشان متوقف شد میتوان به مولکول دارویی RG8039 (یا کوینازولین ۴۹۵) اشاره کرد که مادهای مشتق از کوینازولین بوده و توسط شرکت دارویی «رپلیژن» تولید و در مارس ۲۰۱۴ میلادی تحت لیسانس فایزر قرار گرفت و ظرف مدت کوتاهی پس از انجام فاز ۱ کارآزمایی بالینی، تولیدش متوقف شد. مولکول دیگر PTK-SMA1 نام داشت که به خانوادهٔ تتراسایکلین ها تعلق داشت و توسط شرکت دارویی «پاراتک» تولید شد و قبل از ورود به کارآزمایی بالینی در سال ۲۰۱۰، تولیدش متوقف گشت. داروی RG7800 ملکولی مشابه با RG7916 بود که توسط Hoffmann-La Roche ساخته شد و در سال ۲۰۱۵ میلادی بر روی بیماران آزمایش شد، اما تولیدش به دلیل اثرات سمی بر روی مدلهای حیوانی، برای همیشه متوقف شد.
پژوهشهای پایه، ترکیبات دیگری را کشف کردهاند که پیرایش متناوب SMN2 را به طور آزمایشگاهی تغییر میدهند که سدیم ارتووانادات و آکلاروبیسین از این دستهاند. اولیگونوکلئوتید های آنتیسنس شبه مورفولینو، با همان اهداف سلولی مشابه با داروی نوسینرسن، تحت بررسیهای سخت و گستردهای از جمله در کالج دانشگاهی لندن ودانشگاه آکسفورد هستند.
فعالسازی ژن SMN2
هدف از این روش، افزایش بیان ژنی SMN2 و در نتیجه، بالابردن سطحِ تولیدِ پروتئین SMN در بدن است.
سالبوتامول خوراکی که یکی از داروهای شناختهشدهٔ درمانِ آسم است، هم در مطالعات آزمایشگاهی و هم در سه کارآزمایی کوچک و محدود بالینی، علاوه بر سودمندیهای بالینیاش بر روی دستگاه تنفس، اثرات بالقوهای در درمان آتروفی عضلانی نخاعی نوع ۲ و ۳ داشتهاست.
چند ترکیب شیمیایی هم، با آنکه در آغاز، اثرات امیدبخشی از خود نشان دادند، اما در کارآزماییهای بالینیِ بعدی نتوانستند اثربخشی خود را نشان دهند:
بوتیراتها (مثلاً بوتیرات سدیم و فنیبوتیرات سدیم) در مطالعات آزمایشگاهی اثرات مفیدی از خود نشان دادند، اما در یک کارآزمایی بالینی بر روی بیماران دارای علائم، کارآمدی خود را اثبات نکردند. یک کارآزمایی بالینی دیگر در سال ۲۰۱۵ میلادی، در نوزادان مبتلا به نوع ۱ و ۲ بیماری انجام شده که نتایج آن هنوز منتشر نشده است.
والپروئیک اسید در مطالعات آغازین در دهههای ۱۹۹۰ و ۲۰۰۰ میلادی به طور گستردهای در آزمایشهای تجربی برای درمان SMA بکار میرفت، چرا که بررسیهای آزمایشگاهی نشان داده بود که اثربخشی متوسطی بر روی آن دارد. با این حال، در کارآزماییهای بالینی بزرگ بعدی، اثربخشی چندانی از خود نشان نداد. برخی دانشمندان میگفتند این دارو بر روی افراد خاصی، اثرگذار است اما تأثیراتش در مبتلایان دیگر، توسط آنزیم ترانس لوکاز اسید چرب متوقف میشود. برخی دیگر از متخصصان بر این باورند که والپروئیک اسید نه تنها اثرات مفیدی ندارد، بلکه باعث تشدید علائم بیماری هم میشود.
هیدروکسی اوره (هیدروکسیکارباماید) در موشها مؤثر بودهاست و بههمین دلیل شرکت دانمارکی Novo Nordisk آن را برای مصارف انسانی تحت بررسی و مطالعه قرار داد، اما در نهایت اثرات سودمندی از آن در کارآزماییهای بالینی بر روی بیماران SMA بدست نیامد..
برخی از ترکیبات شیمیایی و داروهایی که به طورآزمایشگاهی فعالیت ژن SMA2 را زیاد کرده اما به مرحله کارآزمایی بالینی نرسیدند، عبارتند از: هورمون رشد ، بازدارندههای هیستون دِ استیلاز، بنزآمید M344، اسیدهای هیدروکسامیک (همچون CBHA و SBHA)، انتینوستات ، پانوبینوستات ، تریکوستاتین A، وُرینوستات، پرولاکتین و همچنین ترکیبات پلی فنل نظیر رسوراترول و کورکومین. داروی سلکوکسیب که یک فعالکنندهٔ مسیر p38-MAPK است، گاهی به صورت خارج از برچسب (off-label) در درمان این بیماری به کار می رود که مبنای آن، تنها یک پژوهش، بر روی مدلهای حیوانی است،اما چنین استفادهای از این دارو، پشتوانهٔ پژوهشی بالینی ندارد.
تهیه شده توسط : ش. سلمانی زاده ( مرکز تحقیقات ژنوم اصفهان – آزمایشگاه ژنتیک پزشکی ژنوم اصفهان )
منبع : NCBI
Add a Comment