





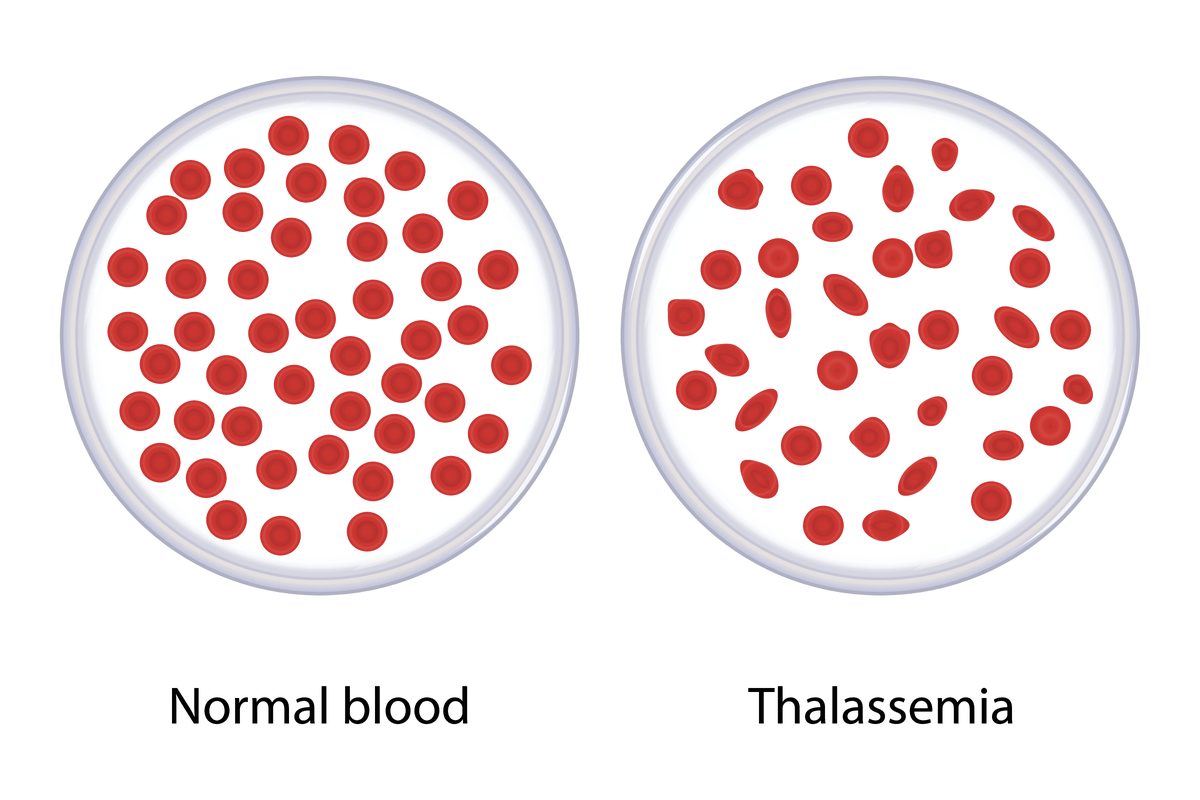
تالاسمی یک نوع اختلال خونی ارثی می باشد که در آن تولید هموگلوبین (پروتئین حامل اکسیژن درگلبول های قرمز خون) کاهش پیدا می کند. این مسئله باعث کمبود گلبول های قرمز در خون و همچنین کاهش سطح اکسیژن در جریان خون و نهایتا طیفی از مشکلات در سلامتی فرد شود.
دو نوع اصلی تالاسمی شامل آلفا تالاسمی و بتا تالاسمی می باشد:
تالاسمی علائم و نشانه های متفاوتی دارد و ممکن است از کم خونی ملایم تا آنمی های شدید، خستگی و بی حالی، زرد شدن رنگ پوست و مشکلات استخوانی متغییر باشد.
بتا تالاسمی با جهش در ژن HBB اتفاق می افتد در حالی که آلفا با جهش در ژن های HBA1 و یا HBA2 اتفاق می افتد. الگوی وراثت تالاسمی اتوزومال مغلوب می باشد. درمان آن وابسته به نوع و شدت بیماری می باشد و ممکن است نیاز به مکمل های اسید فولیک و یا حتی دریافت خون باشد.
علائم و نشانه های تالاسمی:
علائم و نشانه های بسیار وابسته به شدت تالاسمی می باشد برای مثال افراد با فرم های خفیف تالاسمی می توانند کم خونی ضعیف داشته باشند یا در کل هیچ علامت یا نشانه ای بروز ندهند. فرم های intermediate تالاسمی می تواند کم خونی mild تا متوسط را نشان دهد و ممکن است با مشکلات دیگر مانند کاهش رشد، تاخیر در بلوغ، مشکلات استخوانی و افزایش سایز طحال همراه باشد. افراد با فرم شدید تالاسمی علاوه بر علائم در فرم intermediate ممکن است تجربه کم خونی بسیار شدید، کم اشتهایی، بی حالی و رنگ پریدگی، ادرار تیره و رنگ زرد چهره و افزایش اندازه قلب و کبد را داشته باشند.
علت ایجاد بیماری:
دو نوع آلفا وبتا، هر کدام بر روی بخش های متفاوت هموگلوبین تاثیر می گذارد. هموگلوبین از دو واحد متفاوت تشکیل شده است: بتا گلوبین و آلفا گلوبین (α2β2)
ژن های HBB ساختار گلوبین بتا و ژن های HBA1 و HBA2 ساختارگلوبین آلفا را می سازد. هر فرد دو کپی از هر کدام از این ژن ها دارد یک کپی از پدر و یک کپی از مادر به ارث می برد. جهش در ژن HBB منجر به کاهش سطح زنجیره گلوبین بتا و نهایتا بتاتالاسمی می گردد. از دست دادن و حذف بعضی یا همه ژن HBA1 و HBA2 باعث کمبود زنجیره گلوبین آلفا و تالاسمی آلفا می گردد.
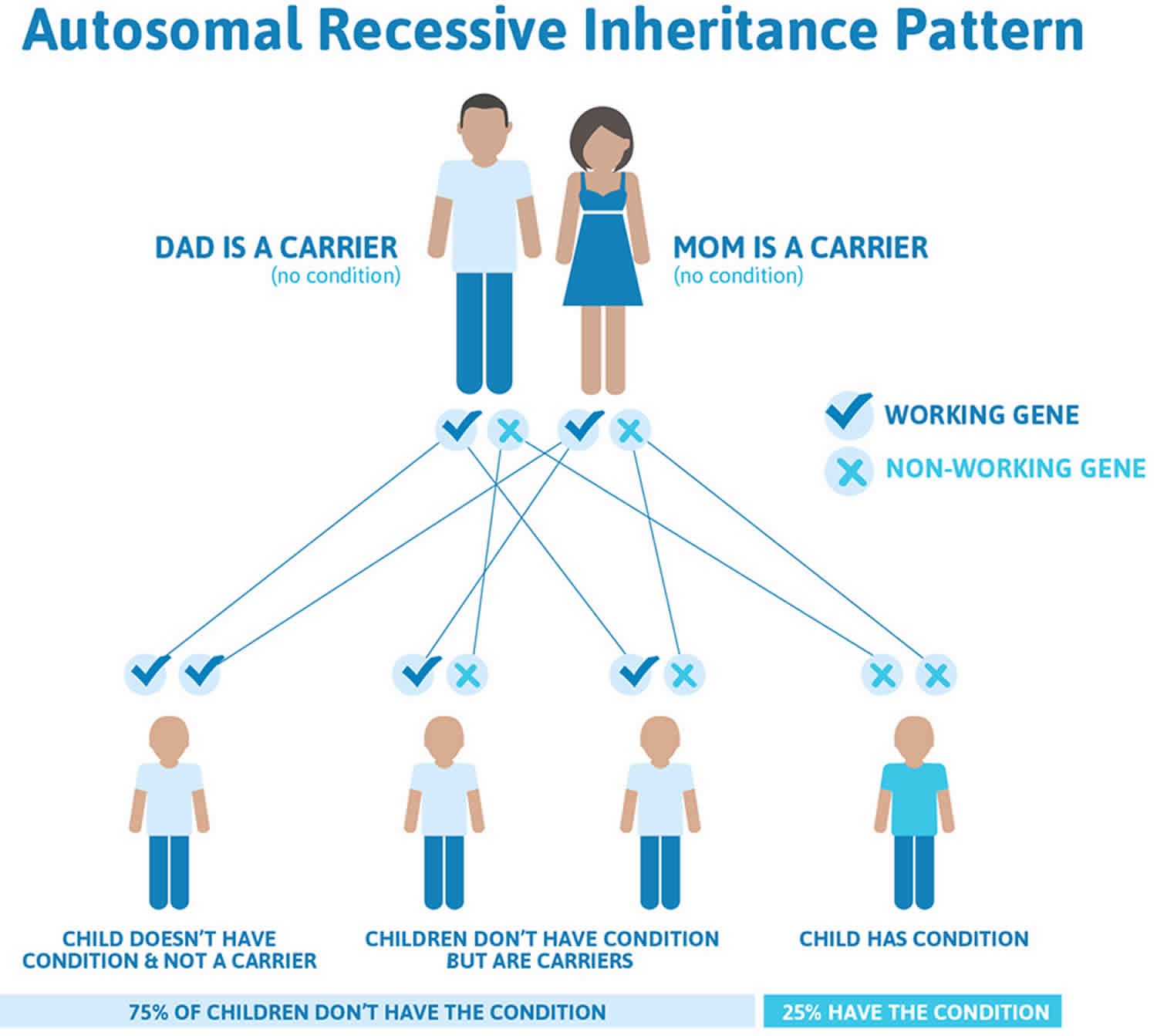
الگوی وراثت:
به طور کلی این بیماری با الگوی وراثت اتوزومال مغلوب به ارث می رسد. بیشتر افرادی که بتا هستند دارای جهش در هر دو کپی ژن HBB در هر سلول می باشند. والدین فرد بیمار معمولا یک کپی جهش یافته از ژن را دارد و به عنوان ناقل یا هتروزیگوت شناخته می شود. ناقلین معمولا علامت یا نشانه ای را ندارند اگرچه بعضی ناقلین بتا، آنمی ملایمی را نشان می دهند. هنگامی که دو ناقل با الگوی وراثت اتوزومال مغلوب بچه دار می شوند هر فرزند با احتمال 25% (1 فرزند از هر 4 فرزند) احتمال تالاسمی ماژور ، 50% احتمال ناقل بودن و 25% شانس نداشتن این بیماری و سالم بودن را دارند.
تالاسمی آلفا بوسیله جهش در دو ژن (HBA1 و HBA2) اتفاق می افتد. افراد دارای دو کپی از HBA1 و دو کپی از HBA2 می باشند برای هر ژن یک کپی از مادر و یک کپی از پدر به ارث می رسد. اگر هر کدام از والدین حداقل در یک کپی از ژن آلفا نقص داشته باشند فرزندان آن ها ریسک به ارث بردن تالاسمی آلفا را دارند. بنابراین، احتمال ابتلا به آلفا تالاسمی و شدت آن بستگی به این دارد که چه تعداد از کپی های ژن آلفا جهش یافته است.
تشخیص:
تست های ژنتیکی برای تشخیص تالاسمی در ژن های HBB وHBA1 ، HBA2 شناخته شده است. اگر جهشی در خانواده ای شناخته شده باشد تست های ناقلی برای اعضای در معرض ریسک خانواده و تشخیص پیش از تولد جنین امکانپذیر می باشد.
در آزمایش خون تالاسمی ماژور گلبولهای قرمز خون کوچک و کم رنگ خواهد بود (کم خونی هیپوکروم میکروسیتر). افت شدید هموگلوبین به مقادیر کمتر از 5 گرم در دسی لیتر وجود دارد. بیلی روبین سرم به علت تخریب سلولها افزایش مییابد. تشخیص قطعی با الکتروفورز هموگلوبین انجام میشود که در تالاسمی ماژور هموگلوبین A طبیعی ساخته نمیشود و 98% هموگلوبینها را هموگلوبین F تشکیل میدهد و هموگلوبین A2 نیز تا 5% افزایش پیدا میکند.
درمان:
انتخاب بهترین روش درمان بر اساس شدت نوع تالاسمی انجام می پذیرد. افراد بیمار با فرم mild اغلب نیاز به درمان خاصی ندارند. درحالی که افراد با تالاسمی intermediate تا فرم شدید آن ممکن است نیاز به تزریق خون، درمان با شلاته کننده های آهن( درمان برای حذف آهن اضافی از بدن بیمار به دلیل سرریز آهن در خون پس از ترزیق ) و مصرف مکمل های اسید فولیک داشته باشند. تالاسمی شدید می تواند باعث مرگ زودهنگام به دلیل مشکلات قلبی گردد در حالی که فرم کمتر شدید آن اغلب طول عمر فرد را کوتاه نمی کند. خوشبختانه انتخاب درمان درست و مناسب باعث افزایش عمر و کیفیت بهتر زندگی برای افرادی با تالاسمی خفیف تا شدید می گردد.
تهیه و ترجمه توسط : خانم ش.سلمانی زاده ( آزمایشگاه ژنتیک پزشکی ژنوم اصفهان – مرکز تحقیقات سلولی و مولکولی و ژنتیک ژنوم )
Add a Comment