




سندرم ماروتو-لامی (موکوپلی ساکاریدوز نوع VI؛ MPS VI) یک اختلال ژنتیکی نادر است که با فقدان کامل یا جزئی فعالیت آنزیم آریل سولفاتاز B (همچنین N-acetylgalactosamine-4-sulfatase) که توسط ژن ARSB کدگذاری میشود، مشخص میشود. کمبود یا عدم فعالیت این آنزیم منجر به تجمع کربوهیدررات پیچیده ای به نام گلیکوزآمینوگلیکان (که قبلاً به عنوان موکوپلی ساکارید شناخته می شد) در بدن می شود.
تجمع غیرطبیعی موکوپلی ساکاریدها منجر به درگیری پیشرونده چندین سیستم اندام می شود. علائم و شدت سندرم Maroteaux-Lamy می تواند به طور چشمگیری از فردی به فرد دیگر متفاوت باشد. برخی از افراد فقط علائم خفیف را ایجاد می کنند، در حالی که برخی دیگر عوارض شدید و حتی تهدید کننده ی زندگی را تجربه می کنند.
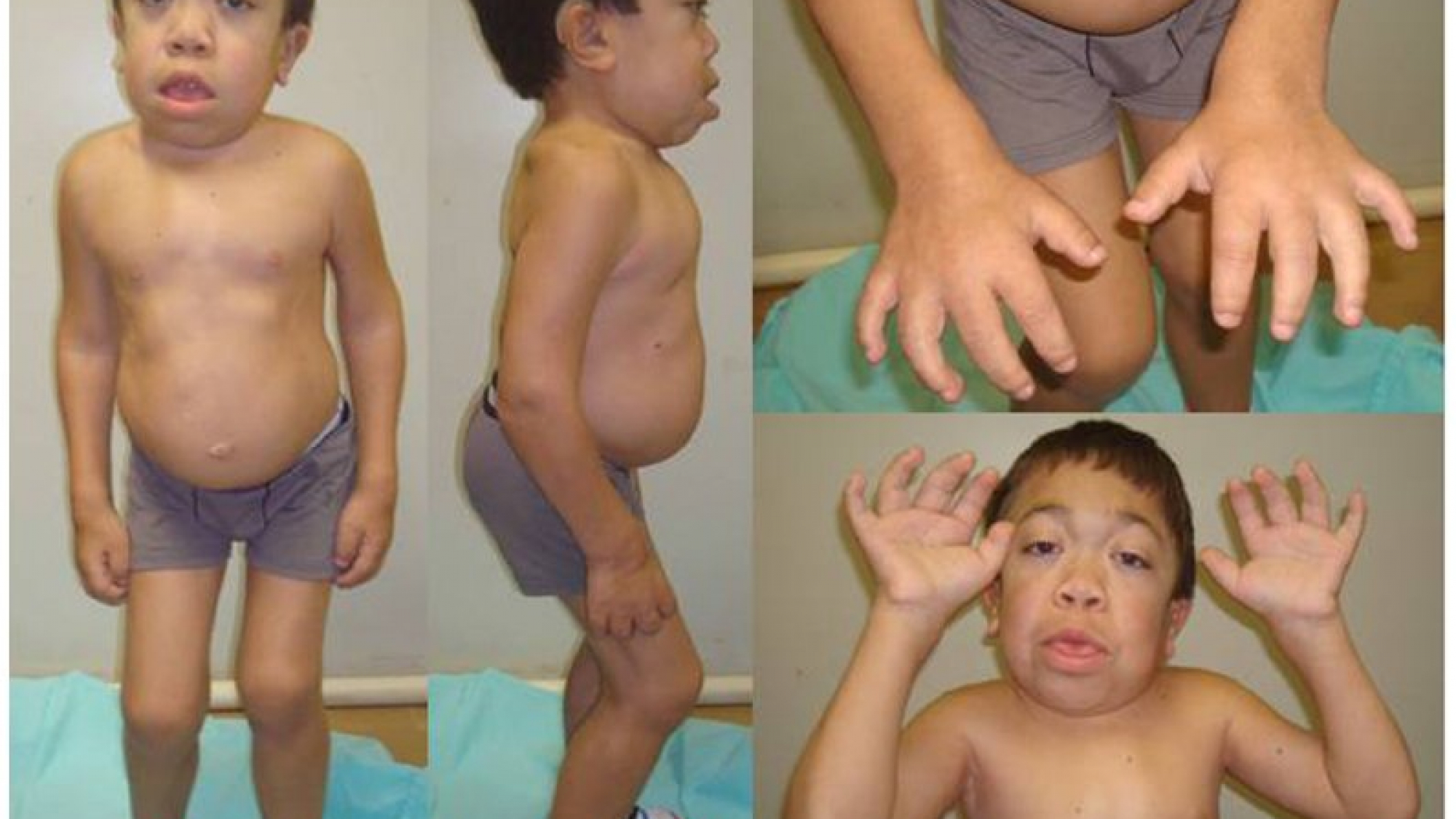
علائم شایع می تواند شامل ویژگی های درشتی صورت، تیرگی قرنیه، ناهنجاری های مفصلی، ناهنجاری های مختلف اسکلتی، بزرگ شدن غیر طبیعی کبد ویا طحال (هپاتواسپلنومگالی) و کاهش شنوایی باشد. بیماری قلبی و بیماری محدود کننده ریوی نیز ممکن است رخ دهد. هوش معمولاً تحت تأثیر قرار نمی گیرد.
موکوپلی ساکاریدوز نوع VI (سندرم ماروتو-لامی)
موکوپلی ساکاریدوز نوع VI (سندرم Maroteaux-Lamy، MPS VI) باعث می شود بدن در تجزیه مولکول های بزرگ قند در سلول ها مشکل داشته باشد. این باعث التهاب و زخم شدن تعدادی از بافت ها و اندام های بدن می شود. علائم می تواند شامل سر بزرگ، تجمع مایع در مغز و سایر اندام ها، تنگ شدن راه های هوایی و کاهش بینایی و شنوایی باشد. MPS VI همچنین می تواند روی اسکلت تأثیر بگذارد.
MPS VI در دوران کودکی شروع میشود و پیشرونده می باشد. متاسفانه افراد مبتلا به MPS VI امید به زندگی کمتری دارند.
ما به عنوان انسان حدود 23000 ژن داریم. این ژن ها مانند دستورالعمل های کوچکی هستند که بر سلامت، رشد و تکامل ما تأثیر می گذارند. ما نیمی از ژن های خود را از مادر بیولوژیکی و نیمی دیگر را از پدر بیولوژیکی خود به ارث می بریم. این ژن ها روی ساختارهایی به نام کروموزوم ها ردیف می شوند. اکثر ما 23 جفت کروموزوم داریم. 22 جفت اول اتوزوم نامیده می شوند و در بیشتر موارد – اینها در بین مردان و زنان یکسان است. جفت 23 جنسیت ما را تعیین می کند – دو کروموزوم X برای یک زن و یک کروموزوم X و یک کروموزوم Y برای مردان.
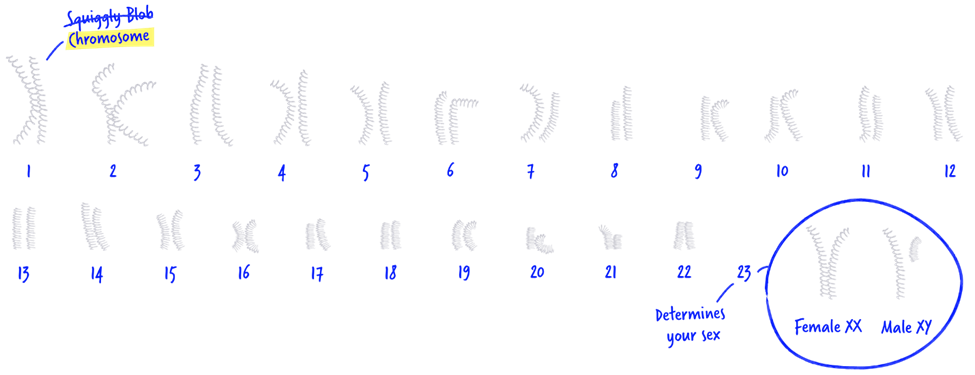
چگونه موکوپلی ساکاریدوز انتقال پیدا میکند؟
موکوپلی ساکاریدوز تیپ 5 به عنوان یک بیماری اتوزومال مغلوب شناخته میشود.برای بیماری اتوزومال اگر فردی تغییری در یک کپی از ژن هایش داشته باشد ناقل است به این معنی که انها ظاهرا سالم هستند زیرا یک کپی از ژنشان فعال است اما میتوانند نسخه ی غیرفعال از ژن معیوب را به فرزندانشان منتقل کنند.
اگر والد دیگر نیز حامل همان ژن معیوب باشند به احتمال 25% فرزند میتواند هردو نسخه ی معیوب را از هردو والد بگیرد و بیمار شود.اگر پدر و مادر هر دو ناقل موکوپلی ساکاریدوز نوع VI (سندرم ماروتو-لامی) باشند، از هر 4 نفر یک نفر احتمال بروز علائم در فرزندانشان وجود دارد.
تشخیص
تشخیص سندرم Maroteaux-Lamy بر اساس شناسایی علائم مشخصه، شرح حال مفصل بیمار و خانواده، ارزیابی بالینی کامل و انواع آزمایشات تخصصی است.
تهیه و ترجمه توسط: خانم شیلا پاکروان ( آزمایشگاه ژنتیک پزشکی ژنوم اصفهان – سیتوژنتیک ).
Add a Comment