





سندروم کریگلر- نجار یک ناهنجاری نادر ژنتیکی است که از طریق ناتوانی در تبدیل صحیح و پاکسازی بیلی روبین از بدن، شناخته می شود.
بیلی روبین یک رنگدانه ی صفراوی نارنجی مایل به زرد است و عمدتا یک محصول جانبی است که از تجزیه طبیعی ( تخریب) گلبول های قرمز قدیمی (همولیز) حاصل می شود. معمولا بیلی روبین تولید شده در این فرآیند، از شکل غیرکونژگه به شکلی تبدیل می شود که می تواند در آب حل شود و از بدن دفع شود( به نام بیلی روبین کونژوگه).
افراد مبتلا به سندروم کریگلر-نجار به علت فقدان آنزیم کبدی خاص مورد نیاز برای تجزیه بیلی روبین، نمی توانند بیلی روبین غیرکونژگه را به فرم کونژوگه تبدیل کنند. از این روی سطوح بالای غیرطبیعی بیلی روبین غیرگونژوگه در خون ( هایپربیلی روبینمی) ایجاد می شود.
علائم بارز سندروم کریگلر-نجار زردی مداوم پوست، غشائ مخاطی و سفیدی چشم( یرقان) است.
دو نوع از این ناهنجاری وجود دارد:
- سندروم کریگلر-نجار نوع I : با کمبود تقریبا کامل فعالیت آنزیم و علائم شدید و حتی تهدید کننده ی زندگی مشخص می شود.
- سندروم کریگلر-نجار نوع II : با فعالیت جزئئ آنزیم و علائم خفیف تر مشخص می شود.
هر دو نوع بیماری، به عنوان صفات اتوزومال مغلوب به ارث می رسند و در اثر جهش در ژن UGT1A1 ایجاد می شوند.
مقدمه
سندروم کریگلر-نجار برای اولین بار در شش نوزاد از سه زوج که دارای رابطه خویشاوندی بودند شناسایی شد. این موارد در تاریخچه پزشکی در سال 1952 توسط دکترها کریگلر و نجار گزارش شدند. در سال 1962، دکتر آریاس یک نسخه ی خفیف تر از این اختلال را گزارش کرد که امروزه به آن سندروم کریگلر-نجار نوع II می گویند.
علائم و نشانه ها
علائم سندروم کریگلر-نجار نوع I ، اندکی پس از تولد آشکار می شود. نوزادان مبتلا دچار زردی شدید و مداوم پوست، غشاهای مخاطی و سفیدی چشم (یرقان) می شوند. این علائم پس از سه هفته ی اول زندگی ادامه می یابد. نوزادان در ماه اول زندگی در معرض خطر ابتلا به کرنیکتروس هستند که به انسفالوپاتی بیلی روبین نیز معروف است.
کرنیکتروس یک وضعیت عصبی تهدید کننده ی زندگی است که در آن مقادیر بالا و سمی بیلی روبین در مغز تجمع یافته و باعث آسیب به سیستم عصبی مرکزی می شود. علائم اولیه کرنیکتروس ممکن اسست شامل کمبود انرژی (بی حالی)، استفراغ، تب و یا تغذیه نامناسب باشد.
علائم دیگری که ممکن است به دنبال داشته باشد عبارتند از: عدم وجود رفلکس های خاص ( رفلکس مورو). اسپاسم عضلانی خفیف تا شدید؛ از جمله اسپاسم هایی که در آن سر و پاشنه پا خم شده یا قوس دار به سمت عقب است. بدن به جلو خم می شود (opisthotonus) و یا حرکات غیر ارادی عضلانی کنترل نشده(spasticity) .
علاوه بر این نوزادان مبتلا ممکن است در شیر خوردن و مکیدن سینه مشکل داشته باشند، با صدای بلند گریه کنند و یا قدرت عضلانی آنها کاهش داشته باشد(هیپوتونی) که منجر به شلی غیرطبیعی بدن می شود.
علائم خفیف
کرنیکتروس می تواند به علائم خفیف تری مانند: کلافگی، مشکل در مهارتهای حرکتی ظریف و توسعه نیافتگی مینای دندان داشته باشد.همچنین به عوارض شدیدی مانند کاهش شنوایی، مشکلات ادراک حسی، تشنج و حرکات آهسته، پیوسته، غیر ارادی و انقباضی (athetosis) بازوها و پاها یا کل بدن منجر شود.
یک دوره کرنیکتروس در نهایت می تواند به آسیب مغزی تهدیدکننده زندگی منجرشود. اگرچه کرنیکتروس معمولا در اوایل دوران نوزادی ایجاد می شود، در برخی موارد، افراد مبتلا به سندروم کریگلر- نجار نوع I ممکن است تا اواخر دوران کودکی یا در اوایل بزرگسالی به کرنیکتروس مبتلا نشوند.
بیمارانی که در آن ها غلظت بیلی روبین خون با قرار گرفتن در معرض نور در سطح ایمن باقی می ماند( تحت درمان قرار دارند). اگر درمان با نور قطع شود یا بیمار تحت تاثیر سایر بیماری ها قرار بگیرد، در هر سنی می توانند به کرنیکتروس مبتلا شوند.
علائم نوع II
سندروم کریگلر- نجار نوع II نسبت به نوع I اختلال خفیف تری است. نوزادان مبتلا دچار یرقان می شوند که در زمان هایی که نوزاد بیمار است ( بیماری هم زمان) و برای مدت طولانی غذا نخورده یا تحت بیهوشی عمومی است با گذشت زمان تشدید می شود. در برخی از افراد، بیماری آن ها تا دوران بزرگسالی تشخیص داده نشده است. کرنیکتروس در کریگلر-نجار نوع II بسیار نادر است. اما می تواند به ویژه در زمانی که فرد مبتلا بیمار است، غذا نخورده یا تحت بیهوشی است رخ دهد.
علل
سندروم کریگلر- نجار در اثر جهش در ژن UGT1A1 ایجاد می شود. ژن ها دستورالعمل هایی را برای ایجاد پروتئین هایی که نقش مهمی را در عملکردهای بدن ایفا می کنند ارائه می دهند. هنگامی که یک جهش در یک ژن رخ می دهد، محصول پروتئینی ممکن است معیوب و ناکارآمد باشد یا اصلا تولید نشود.
بسته به عملکرد ویژه ی آن پروتئین، می تواند بر بسیاری از اندام های بدن از جمله مغز تاثیر بگذارد. ژن UGT1A1 شامل دستورالعمل هایی برای ساخت یک آنزیم کبدی به نام یوریدین دی فسفات گلوکورونوزیل ترانسفراز (UGT1A1) است. این آنزیم برای تبدیل بیلی روبین غیر کونژوگه به بیلی روبین کونژوگه و دفع آن از بدن مورد نیاز است.
علائم در اثر فقدان کامل یا جزئی این آنزیم ناشی می شود که به تجمع بیلی روبین غیر کونژوگه در بدن منجر می شود. بیلی روبین در پلاسمای خون همراه با پروتئینی به نام آلبومین در گردش است، این نوع بیلی روبین، غیرکونژوگه نامیده می شود که در آب حل نمی شود(نامحلول در آب).
به طور طبیعی این بیلی روبین غیر کونژوگه توسط سلول های کبد جذب شده و با کمک آنزیم UGT1A1 به فرم گلوکورونیدهای بیلی روبین (بیلی روبین کونژوگه) که محلول در آب است تبدیل می شوند و در نهایت به صفرا ریخته می شوند.
صفرا در داخل کیسه ی صفرا ذخیره می شود و هنگامی که لازم باشد به مجرای صفراوی مشترک و سپس به قسمت فوقانی روده ی کوچک می ریزد و به هضم کمک می کند. بیشتر بیلی روبین از طریق مدفوع از بدن دفع می شود.
بیشتر شدن سطح بیلی روبین
هنگامی که سطح بیلی روبین به اندازه ی کافی بالا برود. در نهایت می تواند از سد خونی مغزی عبور کند، به بافت مغز نفوذ کرده و باعث بروز علائم عصبی بشود که گاهی با سندروم کریگلر-نجار همراه است. والدین کودکان مبتلا به سندروم کریگلر-نجار نوع I ممکن است برخی نقایص در متابولیسم بیلی روبین را نشان دهند. با این حال هیچ یافته فیزیکی از این اختلال در آن ها مشاهده نمی شود. زیرا فقط یک نسخه از ژن تغییر یافته UGT1A1 (هتروزیگوت) را دارا می باشند، سندروم کریگلر-نجار به صورت اتوزمال مغلوب به ارث می رسد.
ژنتیک
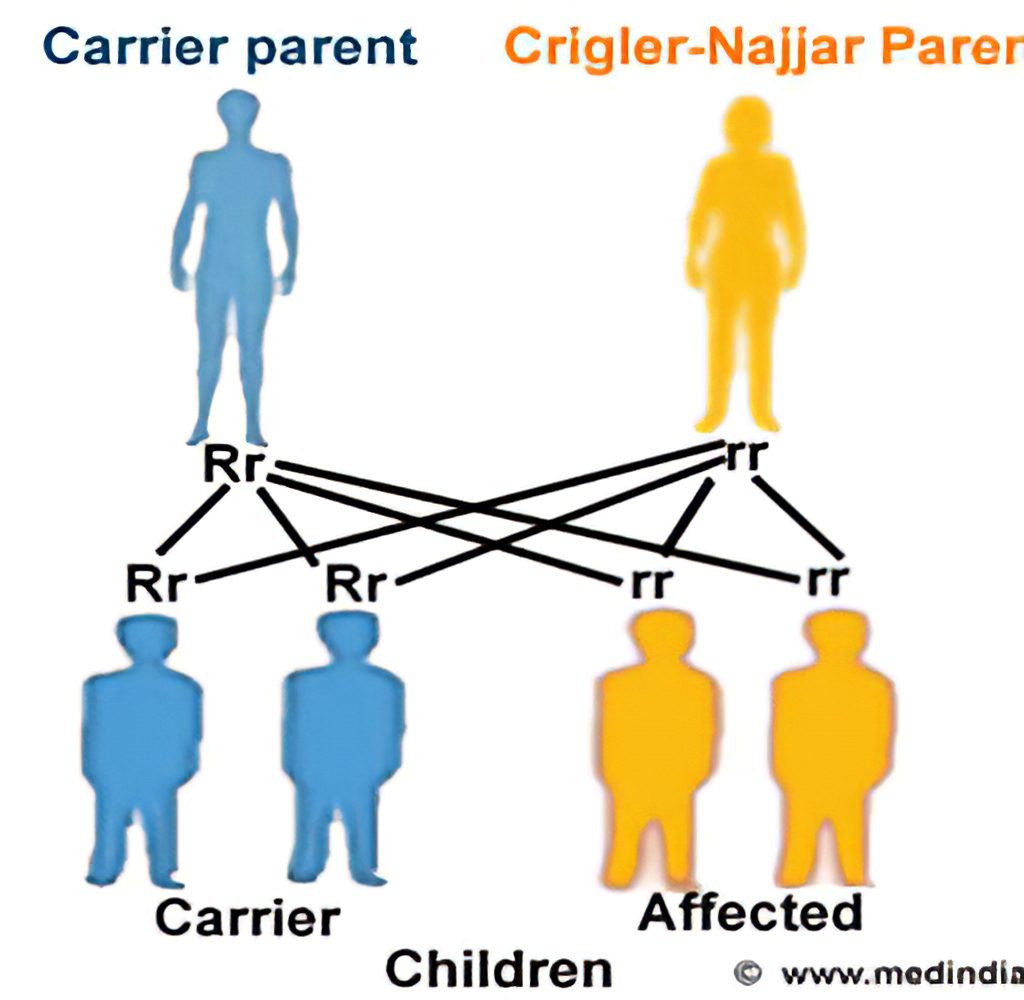
بیماری های ژنتیکی با ترکیب ژن های یک صفت خاص که روی کروموزوم های دریافتی از پدر و مادر هستند تعیین می شوند. بیماری های ژنتیکی مغلوب زمانی رخ می دهند که یک فرد دو کپی از یک ژن غیرطبیعی را برای یک صفت به ارث ببرد(یک کپی از هر والد).
اگر فردی یک ژن طبیعی و یک ژن بیماری زا را به ارث ببرد، آن فرد ناقل بیماری بوده اما معمولا علائمی ندارد. برای والدینی که هر دو ناقل ژن بیماری زا هستند خطر داشتن فرزند بیمار در هر بارداری حدود 25 درصد، احتمال داشتن فرزندی که همانند والدینش ناقل باشد در هر بارداری حدود 50 درصد و همچنین احتمال داشتن فرزندی که هر دو ژن طبیعی را از والدینش دریافت کرده باشد در هر بارداری حدود 25 درصد می باشد. این احتمالات برای جنین دختر و پسر یکسان است.
والدینی که از خویشاوندان نزدیک هستند نسبت به والدین غیرخویشاوندی که هر دو ناقل یک ژن غیر طبیعی هستند احتمال بیشتری برای داشتن فرزندی با اختلال ژنتیکی مغلوب دارند.
جمعیت های هدف
سندروم کریگلر-نجار هر دو جنس را به یک میزان تحت تاثیر قرار می دهد. براساس شواهد میزان بروز در جمعیت عمومی 1 نفر در 750000-1000000 نفر تخمین زده می شود. بسیاری از محققان بر این باور هستند که این اختلال اغلب تشخیص داده نمی شود یا به اشتباه تشخیص داده می شود و تعیین فراوانی واقعی آن در جمعیت عمومی دشوار است. به احتمال زیاد شایع تر از آن چیزی است که تشخیص داده می شود.
تشخیص
در نوزادان مبتلا به زردی مداوم، در چند روز اول زندگی ممکن است به تشخیص این سندروم مشکوک شوند. تشخیص ممکن است با ارزیابی بالینی کامل، یافته های مشخص، شرح حال دقیق بیمار و آزمایش های تخصصی تایید شود. به عنوان مثال، در نوزادان مبتلا به این اختلال، آزمایش خون، سطوح غیر طبیعی بیلی روبین غیرگونژوگه را نشان می دهد.
به علاوه، تجزیه و تحلیل صفرا نشان می دهد که گلوکورونیدهای بیلی روبین قابل تشخیص، وجود ندارد و تجزیه و تحلیل ادرار ممکن است کمبود بیلی روبین را نشان دهد.
آزمایش ژنتیک مولکولی می تواند تشخیص سندروم کریگلر-نجار را تایید کند. آزمایش ژنتیک مولکولی می تواند جهش هایی را در ژن UGT1A1 که به عنوان عامل ایجاد این اختلال شناخته می شوند، شناسایی کند، اما فقط به عنوان یک سرویس تشخیصی در آزمایشگاه های تخصصی در دسترس است. تشخیص سندروم کریگلر-نجار نوع I و نوع II مهم است.
تجویز فنوباربیتال، نوعی باربیتورات، سطح بیلی روبین خون افراد مبتلا به سندروم کریگلر-نجار نوع II و سندروم گیلبرت را کاهش می دهد. اما برای مبتلایان به سندروم کریگلر-نجار نوع I بی اثر است. بنابراین، عدم پاسخ به این دارو نشانه ی مهمی برای اهداف تشخیص افتراقی است.
درمان های استاندارد
این درمان به منظورکاهش سطح بیلی روبین غیرکونژوگه در خون است. درمان زودهنگام در سندروم کریگلر-نجار نوع I برای جلوگیری از ایجاد کرنیکتروس در چند ماه اول زندگی ضروری است. از آنجایی که سندروم کریگلر-نجار نوع II خفیف تر است و به فنوباربیتال پاسخ می دهد، درمان آن متفاوت است.
درمان نوع I
درمان اصلی سندروم کریگلر-نجار نوع I ، فتوتراپی تهاجمی است. در طی این روش، پوست برهنه در معرض نور شدید قرار می گیرد، در حالی که چشم ها محافظت می شوند. این مسئله به تغییر مولکولهای بیلی روبین در پوست کمک می کند، به طوری که می توان آن را در صفرا بدون کونژوگه دفع کرد.
با افزایش سن افراد مبتلا، توده بدن افزایش می یابد و پوست ضخیم می شود.در نتیجه فتوتراپی برای پیشگیری از کرنیکتروس موثر نیست. سال هاست که از نورفلورسنت استفاده می شود اما دارای معایبی ازجمله قرارگرفتن بیماران در معرض اشعه ی فرا بنفش است.
برخی از پزشکان استفاده از فناوری دیودهای ساطع نور(LED) را توصیه می کنند که از نور آبی استفاده می شود. این فناوری را می توان با سطح درمانی خاص مورد نیاز در یک فرد تنظیم کرد و افراد را در معرض اشعه فرابنفش قرار نداد.
با این حال به طور گسترده در دسترس نیست. قرار گرفتن در معرض نور خورشید در کاهش سطح بیلی روبین خون بسیار موثر است. عفونت ها، دوره های تب و سایر انواع بیماری ها، باید فورا درمان شوند تا خطر ابتلای فرد به کرنیکتروس کاهش یابد.
درمان با پلاسما فرزیس
پلاسما فرزیس(plasma pherersis) برای کاهش سریع سطح بیلی روبین در خون استفاده می شود. پلاسما فرزیس روشی برای حذف مواد ناخواسته ( سموم، موادمتابولیک و اجزا ء پلاسما) از خون است. در طی این روش، خون از فرد مبتلا گرفته می شود، سلول های خونی از پلاسما جدا شده و سپس پلاسمای بیمار با پلاسمای انسانی دیگر جایگزین شده و مجددا خون به فرد بیمار تزریق می شود.
پیوند کبد تنها درمان قطعی برای افراد مبتلا به سندروم کریگلر-نجار نوع I است. پیوند کبد دارای معایبی مانند هزینه، محدودیت در دسترس بودن اهدا کننده، نیاز به استفاده ی طولانی مدت از داروهای سرکوب کننده ی سیستم ایمنی و احتمال رد آن است.
درمان در نوزادان و کودکان
در صورتی که نوزادان یا کودکانی که سطح بیلی روبین غیرکونژوگه بالایی دارند به سایر درمان ها پاسخ ندهند( هیپربیلی روبینمی مقاوم به درمان) یا اگر علائم عصبی پیشرفت کند، برخی از پزشکان پیوند کبد را توصیه می کنند. سایر پزشکان معتقدند که پیوند کبد باید قبل از نوجوانی به عنوان درمان پیشگیرانه انجام شود، قبل از اینکه شروع زودرس کرنیکتروس به مغز آسیب بزند.
سندروم کریگلر- نجار نوع II به درمان با فنوباربیتال پاسخ می دهد. در برخی موارد، در طول یک دوره هیپربیلی روبینمی شدید، افراد مبتلا به سندروم کریگلر- نجار نوع II ممکن است به فتوتراپی نیاز داشته باشند. همچنین در برخی از افراد مبتلا ممکن است به هیچ درمانی نیاز نباشد و فقط باید به طور منظم تحت نظر باشند.
مشاوره ژنتیک برای افراد مبتلا و خانواده های آ نها توصیه می شود. حمایت روانی و اجتماعی برای کل خانواده نیز ضروری است.
تهیه و ترجمه توسط : خانم ها شیرین خدابخشیان و پگاه صالحیان ( مرکز ژنتیک پزشکی ژنوم اصفهان – سیتوژنتیک ).
Add a Comment