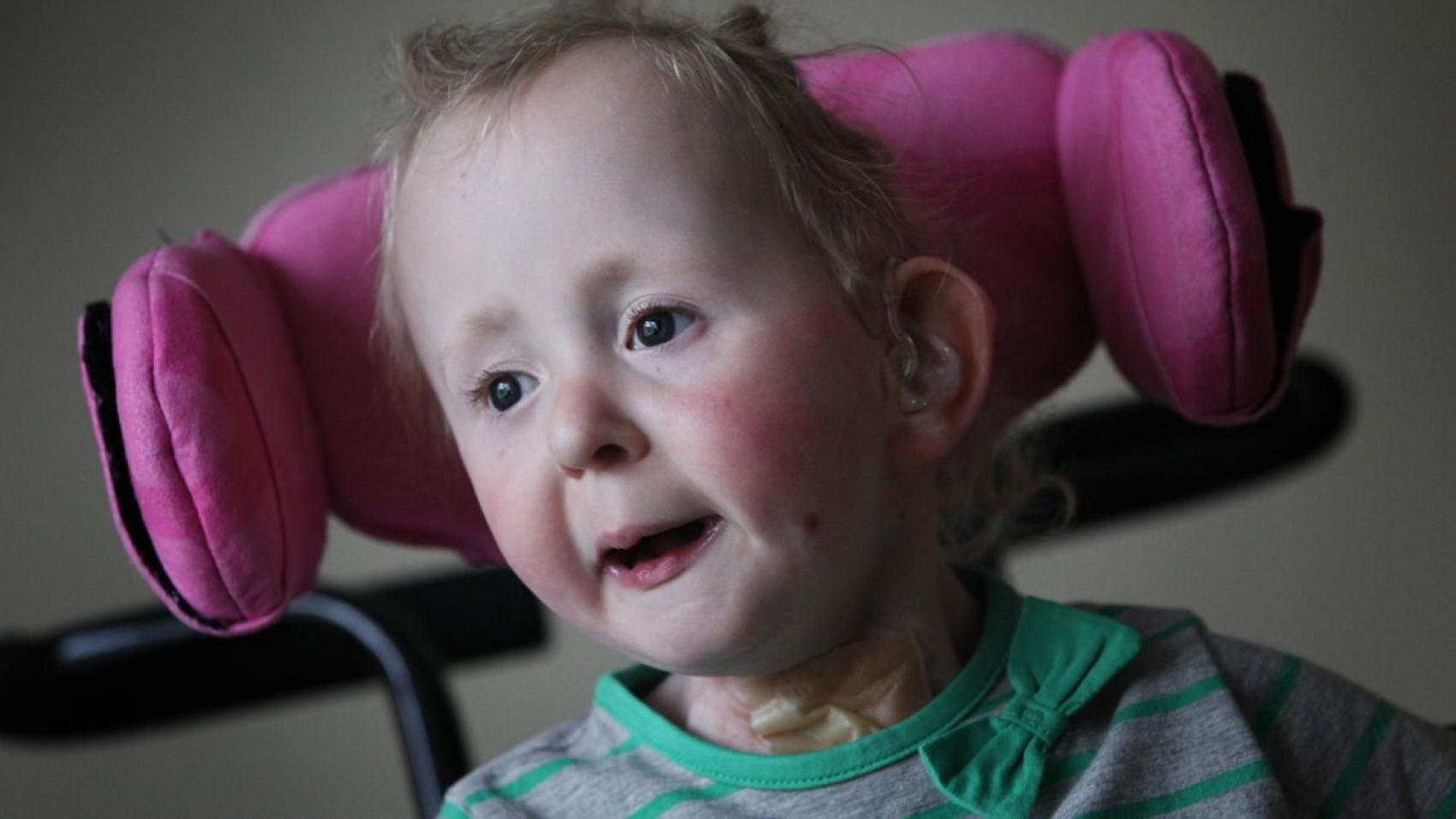
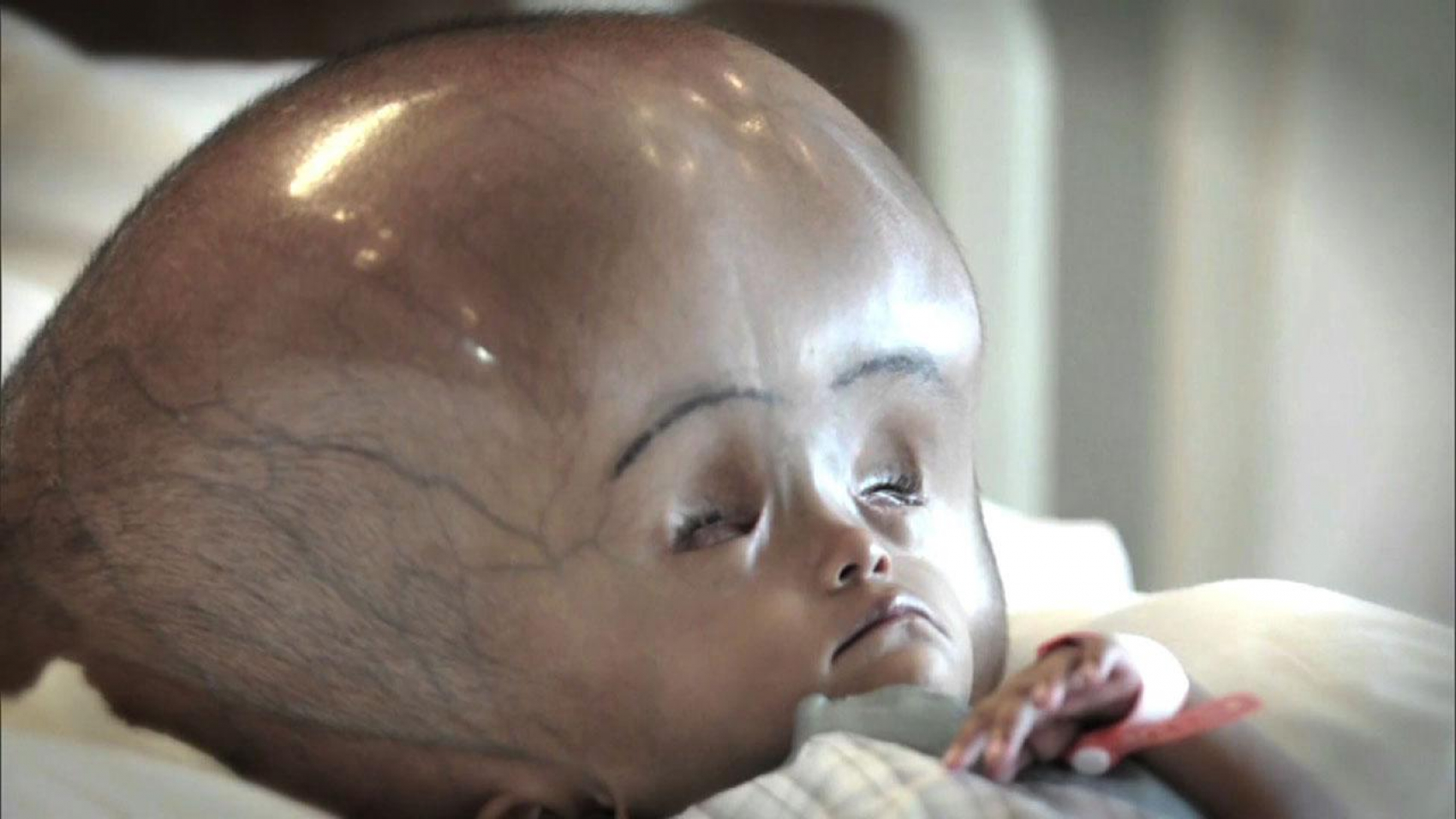
بیماری تای ساکس (TAY-SACHS) یک بیماری نادر است که از والدین به فرزند منتقل می شود. علت آن عدم وجود آنزیمی است که به تجزیه مواد چربی کمک می کند. این مواد چرب که گانگلیوزید نامیده می شوند ، تا سطح سمی در مغز کودک ایجاد شده و بر عملکرد سلول های عصبی تأثیر می گذارد. با پیشرفت بیماری ، کودک کنترل عضلات خود را از دست می دهد. در نهایت ، این بیماری منجر به نابینایی ، فلج و مرگ می شود.