دیستروفی ماهیچه ای امری دریفوس
دیستروفی ماهیچه ای امری دریفوس کمیاب است.این اختلال ژنتیکی که به صورت کند پیش می رود و ماهیچه های بازو,پاها,صورت,گردن,ستون فقرات و قلب را تحت تاثیر قرار می دهد.
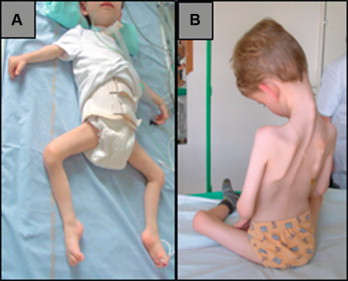
این اختلال شامل ضعف و آتروفی در ماهیچه های مشخص, مفصل هایی که در حالت انعطاف و فشردگی نقش دارند و روی قلب (کاردیومیوپاتی) می شود. علایم اصلی شامل تحلیل و ضعف عضله که منحصرا در بازوها و پایین پاها و انقباض آرنج,تاندون آشیل و ماهیچه های کمر است, می شود.
در تعدادی از موارد اختلالات اضافی ممکن است ظاهر شود. در بیشتر موارد بیماری(EDMD) ارثی و نوع توارث وابسته به جنس یا اتوزومال غالب می باشد. در تعدادی موارد شدیدا کمیاب توارث اتوزومال مغلوب نیز گزارش شده است. اگرچه(EDMD) توارث مختلفی دارد ولی علایم تقریبا مشابهی دارد.
معرفی
(EDMD) به یک گروه از اختلال های ماهیچه ای ژنتیکی کمیاب تحت عنوان دیستروفی عضلانی تعلق میگیرد. مشخصه ی این اختلالات ضعف و آتروفی در ماهیچه های مختلف بدن می باشد. تقریبا 30 اختلال متفاوت دیستروفی عضلانی معرفی شده است. این اختلالات در ماهیچه های متفاوت و سن متفاوت و الگوی توارث و وخامت متفاوتی دارند.
علایم و نشانه ها
سن شروع, وخامت و پیشرفت انواع (EDMD) از موردی به مورد دیگر حتی در بین اعضای یک خانواده متفاوت است. تعدادی از افراد ممکن است در دوران کودکی با پیشرفت سریع بیماری و وخامت پیچیده مبتلا شوند و تعدادی ممکن است در بزرگسالی با پیشرفت اهسته ی بیماری مبتلا شوند.
(EDMD) همراه با ویژگی های بالینی شامل انقباض, ضعف ماهیچه ای و بیماری قلبی می باشد. انقباض در بافت های کوتاه و نازک اتفاق می افتد که باعث دفورمیتی و محدود شدن حرکت در آن ناحیه مخصوصا مفاصل میشود.
آرنج و تاندون آشیل مناطق رایج انقباض می باشند. انقباض گاهی اوقات اولین علایم توارث وابسته به جنس (EDMD) است و ممکن است در اوایل کودکی رخ دهد. در توارث اتوزومال غالب (EDMD) انقباض معمولا بعد از ضعف عضلانی رخ می دهد.
ضعف عضلانی
ضعف عضلانی پیشرفته و تحلیل عضلانی(آتروفی) معمولا در اواخر کودکی یا اوایل بزرگسالی در پشت بازو و پایین پاها رخ می دهد. ضعف و آتروفی ماهیچه ی پاها ممکن است باعث شود راه رفتن کودک تحت تاثیر قرار گیرد و بر روی انگشتان راه برود و به صورت غیرعادی گام بردارد.ضعف ماهیچه ی بازو ممکن است باعث مشکلات متفاوتی شامل سخت کشیده شدن بازوها به بالای سر شود.
در نهایت درگیری ماهیچه های ران و لگن ممکن است صعود از پله را دشوار کنند. و درگیری گردن, شانه, و ساعد باعث سفتی در ستون فقرات شوند. با افزایش سن در افراد مبتلا ممکن است محدودیت در حرکت گردن را تجربه کنند. همچنین ضعف خفیف عضلات صورت نیز گزارش شده است انحنای غیرعادی ستون فقرات (اسکولیوز) نیز ممکن است رخ دهد. ضعف و آتروفی عضلات معمولاً در سه دهه اول زندگی به تدریج پیشرفت می کند. در نهایت ، ممکن است سریعتر شود. برخی از افراد مبتلا به EDMD اتوزومال غالب ممکن است در نهایت توانایی راه رفتن را از دست بدهند و به ویلچر نیاز داشته باشند. ناهنجاری های قلبی سومین ویژگی بارز EDMD هستند و ممکن است منجر به عوارض جدی شود. اگرچه شروع می تواند متفاوت باشد ، ناهنجاری های قلبی معمولاً پس از دهه دوم زندگی ایجاد می شوند.
دیگر علائم
. افراد مبتلا ممکن است به بیماری عضلات قلب (کاردیومیوپاتی) مبتلا شوند که به طور بالقوه منجر به تپش قلب ، خستگی ، ناتوانایی در ورزش و اختلال در توانایی قلب در پمپاژ خون می شود. برخی از افراد ممکن است دچار نقص هایی که منجر به نامنظم شدن ضربان قلب (آریتمی) یا انسداد قلب است, شوند.
انسداد قلب با تداخل در انتقال تکانه های عصبی الکتریکی مشخص می شود که تنظیم کننده ی عملکرد طبیعی ، موزون و پمپاژ عضله قلب می باشد. قلب معمولی دارای چهار حفره است,دو حفره ی فوقانی دهلیز و دو حفره ی پایینی بطن می باشد.در دهلیز راست قلب نرمال یک ضربان ساز طبیعی وجود دارد که ضربان قلب را آغاز و کنترل میکند.
محرک الکتریکی
محرک الکتریکی از ضربان ساز (سینوآتریال یا گره SA )به بطن ها در امتداد یک مسیر بسیار خاص متشکل از بافت رسانا منتقل می شود و به عنوان گره AV( دهلیزی – بطنی) شناخته می شود. تا زمانی که ضربان الکتریکی به طور عادی منتقل شود ، قلب به طور نرمال رفتار می کند,اگر مانع انتقال سیگنال شود ، انتقال مسدود شده به عنوان انسداد قلب یا انسداد AVشناخته می شود.
درجه بندی انسداد قلب
انسداد قلب با توجه به درجه اختلال طبقه بندی می شوند. شدت ناهنجاری های هدایتی در افراد مبتلا به EDMD متفاوت است.و در شکل خفیف انسداد قلب ، دو حفره فوقانی قلب (دهلیز) به طور طبیعی می تپند ، اما انقباضات دو حفره تحتانی (بطن) کمی به تاخیر می افتد. در اشکال شدیدتر ، فقط نیم تا یک چهارم ضربان دهلیزی به بطن ها منتقل می شود در انسداد کامل قلب, دهلیزها و بطنها جداگانه میتپند.در برخی موارد ، انسداد قلب ممکن است منجر به سنکوپ، تنگی نفس و یا ضربان نامنظم قلب (آریتمی) شوند. در موارد شدید ، مرگ ناگهانی امکان پذیر است.
علت ها
در بیشتر موارد (EDMD) توارث وابسته به جنس مغلوب دارد. (EDMD) همچنین ممکن است اتوزومال غالب به ارث برسد. توارث اتوزومال مغلوب بسیار کمیاب می باشد ,اما حداقل در یک خانواده گزارش شده است. بیماری های ژنتیکی با ترکیب ژن ها که روی کروموزوم های دریافت شده از پدر و مادر هستند ، تعیین می شوند.
کروموزوم ها
کروموزوم ها ، که در هسته سلول های انسان وجود دارند ، اطلاعات ژنتیکی را برای هر فرد حمل می کنند, سلولهای بدن انسان به طور معمول دارای 46 کروموزوم هستند. جفت کروموزومهای انسان از 1 تا 22 شماره گذاری می شوند و کروموزومهای جنسی X و Y نام گذاری می شوند. مردها دارای یک کروموزوم X و یک Y و زن ها دارای دو کروموزوم X هستند. هر کروموزوم دارای یک بازوی کوتاه “p” و یک بازوی بلند “q” است. کروموزوم ها به باند های متعددی تقسیم می شوند. به عنوان مثال ، “کروموزوم Xq28” به باند 28 در بازوی بلند کروموزوم X اشاره دارد. باند های شماره گذاری شده محل هزاران ژنی را که در هر کروموزوم وجود دارد ، مشخص می کند.
اختلالات ژنتیکی
اختلالات ژنتیکی وابسته به جنس توسط یک ژن غیر طبیعی روی کروموزوم X ایجاد می شوند و بیشتر در مردان ظاهر می شوند. زنانی که دارای ژن معیوب در یکی از کروموزوم های X خود هستند ، ناقل این اختلال هستند.زنان حامل معمولاً علائم را نشان نمی دهند. زیرا زنان دارای دو کروموزوم X هستند و تنها یکی حامل ژن معیوب است. مرد ها دارای یک کروموزوم X هستند که از مادرشان به ارث رسیده است و اگر یک مرد کروموزوم X را که حاوی ژن معیوب است به ارث ببرد ، به این بیماری مبتلا می شود.
زنان ناقل اختلالات وابسته به جنس در هر بارداری 25٪ شانس داشتن دختر ناقل ، 25٪ شانس داشتن دختر سالم ، 25٪ شانس داشتن پسر مبتلا به این بیماری و 25% شانس داشتن پسری سالم را دارند .اگر یک مرد مبتلا به اختلال وابسته به جنس,قادر به تولید مثل باشد ، ژن معیوب را به تمام دخترانش منتقل می کند.یک مرد نمی تواند ژن مرتبط با X را به پسران خود منتقل کند ، زیرا مردها همیشه کروموزوم Y خود را به جای کروموزوم X خود به فرزندان پسر منتقل می کنند.
جهش ژن
محققان تشخیص داده اند که EDMD وابسته به جنس در اثر جهش ژن EMD که به STA نیز معروف است در بازوی بلند کروموزوم (Xq28)X واقع شده است, رخ می دهد. ژن EMD یک پروتئین ماهیچه ای به نام emerin را کد می کند. امرین در بیشتر سلول های بدن یافت می شود و در ماهیچه های اسکلتی و قلبی دارای سطح بیان بالایی می باشد.
اختلالات ژنتیکی غالب
اختلالات ژنتیکی غالب زمانی رخ می دهد که تنها یک نسخه از ژن غیر طبیعی برای ایجاد یک بیماری خاص لازم است. ژن غیر طبیعی می تواند از هر یک از والدین به ارث برده شود یا می تواند نتیجه جهش جدید در فرد مبتلا باشد. خطر انتقال ژن غیر طبیعی از والدین مبتلا به فرزندان در هر بارداری 50 درصد است. خطر برای مردان و زنان یکسان است.
اختلالات ژنتیکی مغلوب
اختلالات ژنتیکی مغلوب زمانی رخ می دهد که فرد دو نسخه از یک ژن غیر طبیعی را برای یک صفت به ارث می برد. اگر فردی یک ژن طبیعی و یک ژن مربوط به بیماری دریافت کند ، فرد ناقل بیماری خواهد بود اما معمولاً علائمی از خود نشان نمی دهد. خطر هر دو والدین ناقل برای انتقال ژن معیوب و داشتن فرزند مبتلا 25٪ در هر بارداری است.
خطر ناقل شدن فرزند مانند والدین خود که ناقل هستند 50 درصد در هر بارداری است. شانس این که کودک ژن های طبیعی را از والدین دریافت کند. و از نظر ژنتیکی برای آن ویژگی خاص طبیعی باشد 25 درصد است. خطر برای مردان و زنان یکسان است.همه ی اشخاص به طور عادی تعدادی ژن غیرعادی دارند .والدینی که رابطه ی خویشاوندی دارند نسبت به والدینی که رابطه ی خویشاوندی ندارند شانس بیشتری برای داشتن ژن های غیرطبیعی مشابه دارند. و همچنین ریسک داشتن فرزند با اختلالات ژنتیکی مغلوب را بیشتر دارند.
محققان تشخیص داده اند که اشکال اتوزوم غالب و اتوزوم مغلوب EDMD ناشی از جهش های همان ژن واقع در بازوی بلند کروموزوم 1 است.(1q21.2). این ژن به عنوان ژن LMNA شناخته می شود. و پروتئین های lamin A و lamin C را کد می کند. جالب است که جهش های موجود در این ژن باعث ایجاد انواع دیگر بیماری های انسانی از جمله دیستروفی عضلانی لیمب گیردل ، کاردیومیوپاتی ، لیپودیستروفی جزئی خانوادگی از نوع Dunnigan ، و بیماری پیری زودرس سندرم هاچینسون-گیلفورد پروجریا می شود.
دیگر جهش ها
EDMD همچنین می تواند ناشی از جهش در پروتئین های پوششی هسته ای نسپرین 1 و 2 باشد. که مستقیماً با امرین نیز در تعامل هستند. جهش در دومین پروتئین های SUN1 و SUN2 ، که کمپلکسی با نسپرین ها را برای اتصال هسته به اسکلت سلولی تشکیل می دهند ، نیز می تواند باعث EDMD شود. این یافته ها نشان می دهد. که اختلال در مجموعه LINC (پیوند بین اسکلت هسته ای و اسکلت سلولی) می تواند به فنوتیپ عضلانی در EDMD کمک کند.
برخی از موارد EDMD به جهش در ژن FHL1 نسبت داده شده است. که همچنین به عنوان LUMA شناخته می شود. (یک پروتئین غشای هسته ای که به امرین متصل می شود). در عین حال ، بیش از نیمی از بیماران EDMD جهش قابل تشخیص در ژنهای فوق ندارند ، نشان می دهد. که ژن ها و جهش های اضافی باید مسئول EDMD باشند. در نتیجه ، تلاش های اساسی برای شناسایی ژن های اضافی که باعث EDMD و مکانیسم بیماری زمینه ای می شوند ، در دست انجام است.
تشخیص
تشخیص EDMD وابسته به جنس بر اساس ارزیابی بالینی کامل ، شرح حال دقیق بیمار ، شناسایی علائم مشخصه (انقباض ، میوپاتی ، نقایص قلبی و غیره) ,مطالعه میکروسکوپی (بیوپسی) بافت آسیب دیده و تخصصی است. آزمایش هایی مانند تشخیص ایمنی و تست ژنتیک مولکولی امکان پذیر است.
از طریق تشخیص ایمنی ، پزشکان می توانند وجود و میزان پروتئین های خاصی مانند امرین را در نمونه های بافتی بدست آمده از افراد مبتلا تعیین کنند. از تکنیک های مختلفی مانند ایمونوفلورسنس یا وسترن بلات می توان استفاده کرد. این آزمایشات شامل استفاده از آنتی بادی های خاصی است که به پروتئین های خاصی واکنش نشان می دهند.
نمونه هایی که از بیوپسی بافت گرفته شده است در معرض این آنتی بادی ها قرار می گیرد. و نتایج می تواند تعیین کند که آیا پروتئین خاصی مانند امرین وجود دارد و میزان آن چقدر است؟ تقریباً در 95 درصد از افراد مبتلا به EDMD وابسته به جنس امرین وجود ندارد. تست ژنتیکی مولکولی شامل بررسی DNAبرای شناسایی یک جهش ژنتیکی خاص است.
تشخیص EDMD اتوزوم غالب یا مغلوب
تشخیص EDMD اتوزوم غالب یا مغلوب بر اساس ارزیابی بالینی کامل ، شرح حال دقیق بیمار ، شناسایی یافته های مشخصه و آزمایش ژنتیک است. تشخیص ایمنی نمی تواند در تشخیص اشکال اتوزومی EDMD کمک کند. زیرا پروتئین های مرتبط ، لامین A و C ، در افراد مبتلا وجود ندارد.با این حال ، مکان یابی نادرست امرین ، یعنی توزیع غیرعادی امرین در داخل سلول ، اغلب می تواند نشان دهنده جهش در لامین A و C باشد.
آزمایش ها
آزمایش های اضافی که ممکن است برای کمک به تشخیص EDMD مورد استفاده قرار گیرد شامل آزمایش خون و آزمایشی است که سلامت ماهیچه ها و اعصاب کنترل کننده ماهیچه ها را ارزیابی می کند. ( الکترومیوگرافی ). آزمایش خون ممکن است سطح بالایی از کراتین کیناز (CK) را نشان دهد. آنزیمی که اغلب هنگام آسیب ماهیچه ها در سطوح غیر طبیعی بالا یافت می شود. تشخیص افزایش سطح CK می تواند آسیب یا ملتهب شدن ماهیچه را تأیید کند ، اما نمی تواند تشخیص EDMD را تأیید کند.
در طی الکترومیوگرافی ، یک الکترود سوزنی از طریق پوست به عضله آسیب دیده وارد می شود.الکترود فعالیت الکتریکی عضله را ثبت می کند.این رکورد نشان می دهد که یک ماهیچه چقدر به اعصاب واکنش نشان می دهد. و می تواند تعیین کند که آیا ضعف عضلانی ناشی از خود عضله است یا عصب های کنترل کننده ماهیچه ها الکترومیوگرافی می تواند اختلالات عصبی مانند بیماری نورون حرکتی و نوروپاتی محیطی را رد کند.
افراد مبتلا به EDMD ممکن است الکتروکاردیوگرام دریافت کنند. آزمایشی که تکانه های الکتریکی قلب را ثبت می کند و ممکن است الگوهای الکتریکی غیر طبیعی را نشان دهد.
درمان
درمان خاصی برای EDMD وجود ندارد.درمان با توجه به علائم خاصی که در هر فرد وجود دارد ، انجام می شود.گزینه های درمانی ممکن است شامل فیزیوتراپی و ورزش برای تقویت قدرت عضلات و جلوگیری از انقباضات باشد. ممکن است در برخی موارد برای درمان انقباضات یا اسکولیوز جراحی توصیه شود. استفاده از وسایل کمکی مکانیکی (مانند عصا ، ویلچر) ممکن است برای کمک به راه رفتن ضروری باشد.