سندرم تریچر کالینز (TCS)

سندرم تریچر کالینز (TCS) یک اختلال مادرزادی جمجمه و صورت است. که با هیپوپلازی (عدم رشد) استخوان گونه و فک پایین و ناهنجاریهای اطراف چشم مشخص می شود. اگرچه نام آن به «ادوارد تريچر کالينز»، چشم پزشک انگليسي برمی گردد. که اين وضعيت را در سال 1900 توصيف کرد، اما توصيف اوليۀ آن توسط «تامسون» در سال 1846 و سپس «بري» در سال 1889 انجام شد.ً در سال 1949، فرانچستي و کلين اين اختلال را بررسي کردند. و اصطلاح «دیسوستوز فک پایین» (سندرم فرانچستی-کلین) را برای این بیماری پیشنهاد کردند.
شرح بیماری
همانطور که در مورد اختلالات نادر اتفاق میافتد، تعداد کمی از استراتژی های درمانی سطح بالا وجود دارند که دیسمورفولوژی TCS را هدف قرار می دهند. با درنظر گرفتن این موضوع، درمان بیماران مبتلا به TCS از اصولی پیروی می کند و شامل مراحل پایۀ جراحی جمجمه و صورت در کنار سایر دستکاریهای استخوانی و سپس بازسازی بافت نرم است.
اولویت با مسائل عملکردی و پس از آن نگرانیهای زیبایی با بلوغ صورت بیماران است. به دلیل پیچیدگی و طیف گستردهای از ناهنجاریها که در این بیماری اتفاق میافتد، درمان کودکان متولد شده با TCS نیاز به متخصصان رشتههای مختلف دارد. ممکن است علاوه بر جراح پلاستیک جمجمه، تخصص متخصصان چشم، گوش، حلق و بینی، آسیبشناسی گفتار، شنواییشناسی، ارتودنسی، ژنتیک، تنفس، اطفال و مراقبتهای ویژه لازم باشد. همچنین، مشاورۀ بیمار و خانواده برای دستیابی به کیفیت مطلوب زندگی از اهمیت اساسی برخوردار است، زیرا این بیماران به احتمال زیاد نیاز به مداخلات متعدد در دوران کودکی و اغلب تا بزرگسالی دارند.
ژن ها
TCS یک اختلال اتوزومال غالب با نفوذپذیری متغیر است. میزان بروز این بیماری، 1 در هر 50000 تولد زنده تخمین زده می شود. جهش در ژنهای TCOF1، POLR1D و POLR1C در این بیماری نقش دارند. اکثر این جهشها در ژن TCOF1 که بر روی کروموزوم 5 ناحیۀ q31.3-q33.3 واقع شده و پروتئین Treacle را کد میکند اتفاق می افتند. جهش در این ژن منجر به نقص در بیوژنز ریبوزوم و نارسایی سلولهای نورالکرست می شود. تعدادی از بیماران مبتلا به این اختلال هیچ جهشی را نشان نمی دهند. .همچنین، مطالعات یک الگوی وراثتی اتوزومال مغلوب را گزارش میکنند (POLR1C). شصت درصد موارد، جهشهای de novo را نشان میدهند و 40 درصد دارای جهش های خاص خانوادگی هستند. هیچ ارتباطی بین فنوتیپ و ژنوتیپ دیده نشده است.

علائم
بیماران متولد شده با این اختلال، از نظر فنوتیپی تنوع گستردهای دارند. در حالی که برخی از بیماران می توانند ناهنجاری خفیف اطراف چشم را که از نظر بالینی نامحسوس است نشان دهند. برخی دیگر فنوتیپ کاملتری را با ناهنجاریهای شدید دور چشم مثل کجی چشمها به سمت پایین، دیستوپی کانتال (جابجایی گوشه داخلی چشم) و کلوبوما (قطرهای بودن مردمک چشم)، هیپوپلازی فک بالا، و تغییر مکان خط رویش مو و اشکال مختلف میکروتیا (بدشکلی گوش خارجی) نشان می دهند.
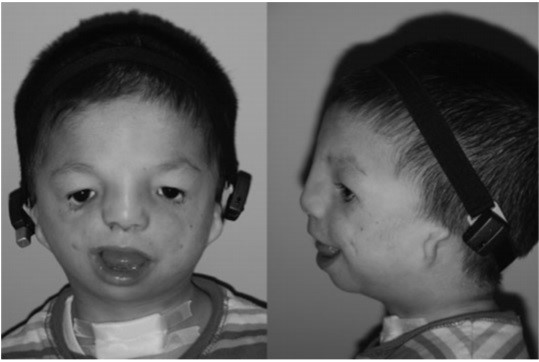
(سمت راست) میکروتیا، بقایای گوش و خط موی جابجا شده. یک باند سمعک که دور استخوان جمجمه پیچیده است در محل وجود دارد.
(سمت چپ) ناهنجاریهای دور چشم شامل کجی به سمت پایین و اکتروپیون (برگشتن لبه پلک (معمولاً پلک پایین). و مژه ها به سمت بیرون چشم) و بزرگ شدن اسکلر (صلبیه).
ویژگی ها
صرف نظر از شدت، تغییر شکل، دو طرفه و بهطور کلی متقارن است. ویژگیهای اصلی این بیماری نشاندهندۀ ناهنجاری اساسی منشأگرفته از قوسهای branchial اول و دوم است. ناهنجاری های دیگر شامل میکروتیا با کاهش شنوایی و تأخیر احتمالی در گفتار، هیپوپلازی فک پایین و رتروگناتیا (تورفتگی چانه) با عوارض احتمالی در راه هوایی و شکاف کام است (در 40 درصد موارد). ناتوانی ذهنی و ناهنجاریهای دیگر مثل ناهنجاریهای قلبی در زمینۀ این جهش ژنتیکی گزارش شده است.
آنالیز ژنتیکی همچنان روش قطعی تشخیص TCS ، قبل یا بعد از تولد است. سونوگرافی قبل از تولد نیز در تشخیص TCS مورد استفاده است. اما این روش با وجود توانایی تشخیص برخی از مشخصههای صورت در TCS، به تنهایی نمیتواند بین سندرمهای مشابه دیسوستوز صورت تمایز قائل شود. علاوه بر این، با توجه به اینکه گروهی از بیماران ممکن است جهش ژنتیکی نداشته باشند. و همینطور با وجود تنوع فنوتیپی و عدم ارتباط بین فنوتیپ و ژنوتیپ، نتایج آزمایش ژنتیک قبل از تولد باید با احتیاط تفسیر شود.