





دیزومی تک والدی چیست؟
به طور طبیعی هر فرد یک کپی از هر جفت کروموزومش را از مادر بیولوژیکی اش و کپی دیگر از همان جفت کروموزوم را از پدر بیولوژیکی اش به ارث میبرد. دیزومی تک والدی به وضعیتی اطلاق میشود. که 2 کپی از یک کروموزوم از یک والد به ارث میرسد. سندروم آنجلمن (AS) و سندروم پرادر-ویلی(PWS) نمونه هایی از اختلالاتی هستند که می توانند دز اثر دیزومی تک والدی ایجاد شوند.
سندروم آنجلمن چیست؟
سندرم آنجلمن یک اختلال ژنتیکی پیچیده است که در درجه اول سیستم عصبی را تحت تاثیر قرار می دهد. ویژگی های مشخص این بیماری شامل تاخیر در رشد ، ناتوانی ذهنی ، اختلال شدید گفتار و مشکلات حرکتی و تعادل (آتاکسی) است. اکثر کودکان مبتلا نیز تشنج های مکرر (صرع) و اندازه سر کوچک (میکروسفالی) دارند. تاخیر در رشد در سن 6 تا 12 ماهگی مشهود می شود. و سایر علائم و نشانه های معمول معمولاً در اوایل کودکی ظاهر می شوند. کودکان مبتلا به سندرم آنجلمن معمولاً دارای رفتار شاد و هیجان انگیز با لبخند مکرر ، خنده و حرکات دست زدن هستند. بیش فعالی ، محدوده توجه کوتاه مدت و شیفتگی به آب معمول است.
ویژگی های رایج
با افزایش سن ،در افراد مبتلا به سندرم آنجلمن تحریک پذیری کاهش پیدا میکند. و مشکلات خواب در این افراد نیز بهبود می یابد. با این حال ، افراد مبتلا همچنان با نقص ذهنی ، نقص شدید گفتار و تشنج به زندگی خود ادامه می دهند. بزرگسالان مبتلا به سندرم آنجلمن دارای ویژگی های متمایز صورت هستند که ممکن است به عنوان “صورت درشت” توصیف شوند. سایر ویژگی های رایج شامل پوست غیرمعمول و روشن با موهای روشن و انحنای غیرطبیعی ستون فقرات (اسکولیوزیس) است. به نظر می رسد امید به زندگی افراد مبتلا به این بیماری تقریباً طبیعی است.
علت ایجاد سندروم آنجلمن:
بسیاری از ویژگی های سندرم آنجلمن ناشی از دست دادن عملکرد ژنی به نام UBE3A است. افراد به طور معمول یک نسخه از ژن UBE3A را از هر یک از والدین به ارث می برند. هردو نسخه از این ژن در بسیاری از بافتهای بدن روشن (فعال) است. با این حال ، در مناطق خاصی از مغز ، فقط نسخه ای که از مادر شخص به ارث رسیده است (نسخه مادری) فعال است. این فعال سازی ژن مختص والدین توسط پدیده ای به نام نقشه گذاری ژنومی (Genomic imprinting) ایجاد می شود.
اگر نسخه مادری ژن UBE3A به دلیل تغییر کروموزومی یا جهش ژنی از بین برود ، فرد هیچ کپی فعال ازاین ژن در برخی از قسمت های مغز نخواهد داشت. چندین مکانیسم ژنتیکی مختلف می توانند نسخه مادری ژن UBE3A را غیرفعال یا حذف کنند.
جهش
دربیشتر موارد سندرم آنجلمن (حدود 70 درصد) زمانی اتفاق می افتد که قسمتی از کروموزوم 15 مادری حاوی این ژن حذف شود. در موارد دیگر (حدود 11 درصد) ، سندرم آنجلمن ناشی از جهش در نسخه مادری ژن UBE3A است. در درصد کمی از موارد ، سندرم آنجلمن زمانی به وجود می آید که فرد دو نسخه از کروموزوم 15 را به جای یک نسخه از هر یک از والدین ، از پدر خود (نسخه های پدری) به ارث می برد. این پدیده دیزومی تک والدی پدری نامیده میشود. به ندرت ، سندرم آنجلمن نیز می تواند در اثر بازآرایی کروموزومی به نام جابجایی یا جهش یا نقص دیگری در ناحیه DNA ایجاد شود که فعال سازی ژن UBE3A را کنترل می کند.
علل سندرم آنجلمن
این تغییرات ژنتیکی می تواند به طور غیرعادی UBE3A یا سایر ژن ها را در نسخه مادری کروموزوم 15 خاموش (غیرفعال) کند. علل سندرم آنجلمن در 10 تا 15 درصد افراد مبتلا ناشناخته است. در این موارد تغییرات مربوط به ژنها یا کروموزومهای دیگر ممکن است عامل این اختلال باشد. در برخی از افراد مبتلا به سندرم آنجلمن ، از دست دادن ژنی به نام OCA2 با موهای روشن و پوست روشن همراه است. ژن OCA2 در قسمتی از کروموزوم 15 قرار دارد که اغلب در افراد مبتلا به این اختلال حذف می شود. با این حال ، از دست دادن ژن OCA2 علائم و نشانه های دیگر سندرم آنجلمن را ایجاد نمی کند. پروتئین تولید شده از این ژن به تعیین رنگ (رنگدانه شدن) پوست ، مو و چشم کمک می کند.
وراثت:
بیشتر موارد سندرم آنجلمن ارثی نیستند . به ویژه مواردی که در اثر یک حذف در کروموزوم 15 مادری یا دیزومی تک والدی پدری ایجاد می شود. این تغییرات ژنتیکی به صورت رویدادهای تصادفی در حین تشکیل سلول های باروری (تخمک و اسپرم) یا در مراحل اولیه رشد جنینی رخ می دهد. افراد مبتلا معمولاً سابقه این ناهنجاری را در خانواده خود ندارند. به ندرت ، یک تغییر ژنتیکی مسئول سندرم آنجلمن می تواند ارثی باشد. به عنوان مثال ، ممکن است جهش در ژن UBE3A یا در ناحیه نزدیک DNA که فعال سازی ژن را کنترل می کند ، از نسلی به نسل دیگر منتقل شود.
تشخیص:
تشخیص اولیه این بیماری توسط پزشک براساس علائم بالینی انجام میشود . در مراحل بعدی جهت تایید علائم، ترکیبی از آزمایشهای ژنتیکی که شامل موارد زیر است صورت میگیرد:
- تجزیه و تحلیل کروموزوم جهت بررسی اندازه، شکل و تعداد کروموزومها در سلول.
- نکنیک FISH (هیبریداسیون فلوئورسنت در محل). جهت تشخیص کروموزوم از دست رفته.
- آزمایش متیلاسیون DNA برای بررسی اینکه آیا هر دو نسخه از یک ژن – یکی از مادر و دیگری از پدر – فعال هستند یا خیر.
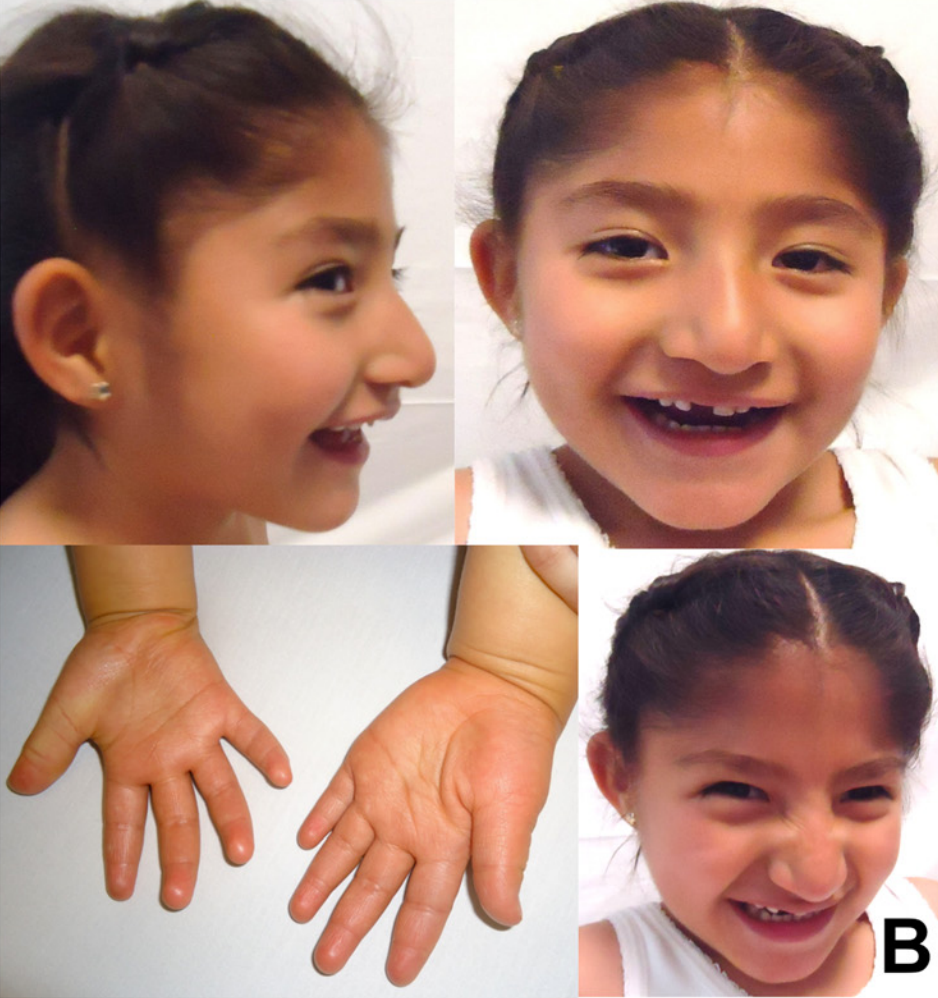
سندروم پرادر-ویلی چیست؟
سندرم پرادر-ویلی یک بیماری ژنتیکی پیچیده است که قسمت های زیادی از بدن را درگیر می کند. در دوران نوزادی ، این اختلال با ضعف عضلانی (هیپوتونی) ، مشکلات تغذیه ، رشد ضعیف و تاخیر در رشد مشخص می شود. از ابتدای کودکی ، افراد مبتلا اشتهای سیری ناپذیری را تجربه می کنند. که منجر به پرخوری مزمن (هایپرفاژیا) و چاقی می شود. برخی از افراد مبتلا به سندرم پرادر-ویلی ، به ویژه افرادی که چاقی دارند ، به دیابت نوع 2 (رایج ترین نوع دیابت) نیز مبتلا می شوند.
نشانه ها
افراد مبتلا به سندرم پرادر-ویلی معمولاً دارای نقص ذهنی خفیف تا متوسط و اختلالات یادگیری هستند. مشکلات رفتاری نظیر سرسختی و رفتارهای اجباری نظیر کندن پوست در این افراد شایع است. همچنین این افراد دارای اختلالات خواب نیز هستند. ویژگیهای ظاهری این بیماری شامل ویژگیهای بارز صورت مانند پیشانی باریک ، چشمهای بادامی شکل و دهان مثلثی و همچنین قد کوتاه ، دست ها و پاهای کوچک میباشد. مردان مبتلا و زنان مبتلا دارای اندام تناسلی توسعه نیافته هستند. بلوغ دیر یا ناقص است و اکثر افراد مبتلا نمی توانند بچه دار شوند (نابارور).
علت ایجاد سندروم پرادر-ویلی:
سندرم پرادر-ویلی در اثر از بین رفتن عملکرد ژن ها در یک ناحیه خاص از کروموزوم 15 ایجاد می شود. افراد به طور معمول از هر والدین خود یک نسخه از این کروموزوم را به ارث می برند. برخی از ژن ها فقط در نسخهای که از پدر (نسخه پدری) روشن (فعال) هستند. این فعال شدن ژن مختص والدی توسط پدیده ای به نام نقشه گذاری ژنومی ایجاد میشود. اکثر موارد سندرم پرادر-ویلی (حدود 70 درصد) زمانی اتفاق می افتد که قسمتی از کروموزوم 15 پدری در هر سلول حذف شود.
ژن ها
افرادی که دچار این تغییر کروموزومی هستند ، ژن های حیاتی خاصی را در این ناحیه از دست می دهند. زیرا ژن های روی نسخه پدر حذف شده اند و ژن های روی نسخه مادر خاموش هستند (غیر فعال). در 25 درصد موارد دیگر ، فرد مبتلا به سندرم پرادر-ویلی به جای یک نسخه از هر والدین ، دو نسخه از کروموزوم 15 را که از مادرش (نسخه مادری) به ارث میبرد. این پدیده دیزومی تک والدی مادری نام دارد. به ندرت ، سندرم پرادر-ویلی همچنین می تواند در اثر بازآرایی کروموزومی به نام انتقال یا در اثر جهش یا نقص دیگری ایجاد شود که به طور غیرعادی ژن های کروموزوم 15 پدری را خاموش (غیرفعال) می کند.
این سندرم ناشی از چیست؟
به نظر می رسد ویژگی های مشخصه سندرم پرادر-ویلی ناشی از از دست دادن عملکرد چندین ژن در کروموزوم 15 است. در میان آنها ژن هایی وجود دارد که دستورالعمل ساخت مولکول هایی به نام RNA های کوچک هسته ای (snoRNAs) را ارائه می دهند. این مولکولها عملکردهای مختلفی دارند ، از جمله کمک به تنظیم انواع دیگر مولکولهای RNA (مولکولهای RNA نقش مهمی را در تولید پروتئین و سایر فعالیتهای سلول ایفا میکنند). مطالعات نشان می دهد که از دست دادن گروه خاصی از ژن های snoRNA ، که به عنوان خوشه SNORD116 شناخته می شوند ، ممکن است نقش عمده ای در ایجاد علائم و نشانه های سندرم پرادر-ویلی داشته باشند.
با این حال ، هنوز مشخص نیست که چگونه یک خوشه SNORD116 مفقود شده می تواند به ناتوانی ذهنی ، مشکلات رفتاری و ویژگی های جسمانی این اختلال کمک کند.
ژن OCA2
در برخی از افراد مبتلا به سندرم پرادر-ویلی ، از دست دادن ژنی به نام OCA2 با پوست غیرمعمول و موهای روشن روشن ارتباط دارد. ژن OCA2 در قسمتی از کروموزوم 15 قرار دارد که اغلب در افراد مبتلا به این اختلال حذف می شود. با این حال ، از دست دادن ژن OCA2 علائم و نشانه های دیگر سندرم پرادر-ویلی را ایجاد نمی کند. پروتئین تولید شده از این ژن به تعیین رنگ (رنگدانه شدن) پوست ، مو و چشم کمک می کند. محققان در حال مطالعه ژن های دیگر روی کروموزوم 15 هستند که ممکن است مربوط به علائم و نشانه های اصلی این بیماری باشد.
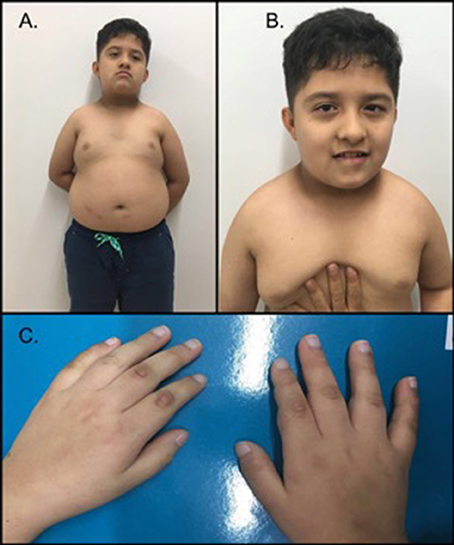
وراثت:
اکثر موارد سندرم پرادر-ویلی ارثی نیستند ، به ویژه مواردی که در اثر حذف در کروموزوم پدری 15 یا در اثر دیزومی تک والدی مادری ایجاد می شود. این تغییرات ژنتیکی به صورت رویدادهای تصادفی در حین تشکیل سلول های باروری (تخمک و اسپرم) یا در مراحل اولیه رشد جنینی رخ می دهد. افراد مبتلا معمولاً سابقه اختلال در خانواده خود را ندارند. به ندرت ، یک تغییر ژنتیکی مسئول سندرم پرادر-ویلی می تواند ارثی باشد. به عنوان مثال ، ممکن است یک تغییر ژنتیکی که ژن های کروموزوم پدری 15 را به طور غیرعادی غیر فعال می کند ، از نسلی به نسل دیگر منتقل شود.
تشخیص:
تشخیص این بیماری در مرحله اول براساس علائم بالینی توسط پزشک انجام میگیرد. در مرحله بعد تشخیص قطعی از طریق آزمایش خون و تستهای ژنتیکی صورت میگیرد. روش ترجیحی، آزمایش “تجزیه و تحلیل متیلاسیون” است که 99٪ موارد ، از جمله همه زیرگونه های اصلی ژنتیکی PWS (حذف ، دیزومی تک والدی ، یا خطای نقشهگذاری) را تشخیص می دهد. آزمایش بعدی FISH” (هیبریداسیون فلوئورسنت در محل) است که بیماران مبتلا به PWS به دلیل حذف ژنی را شناسایی می کند ، اما افراد مبتلا به سندرم پرادر-ویلی به دلیل دیزومی تک والدی یا خطای نقشهگذاری را مشخص نمی کند.
Add a Comment