


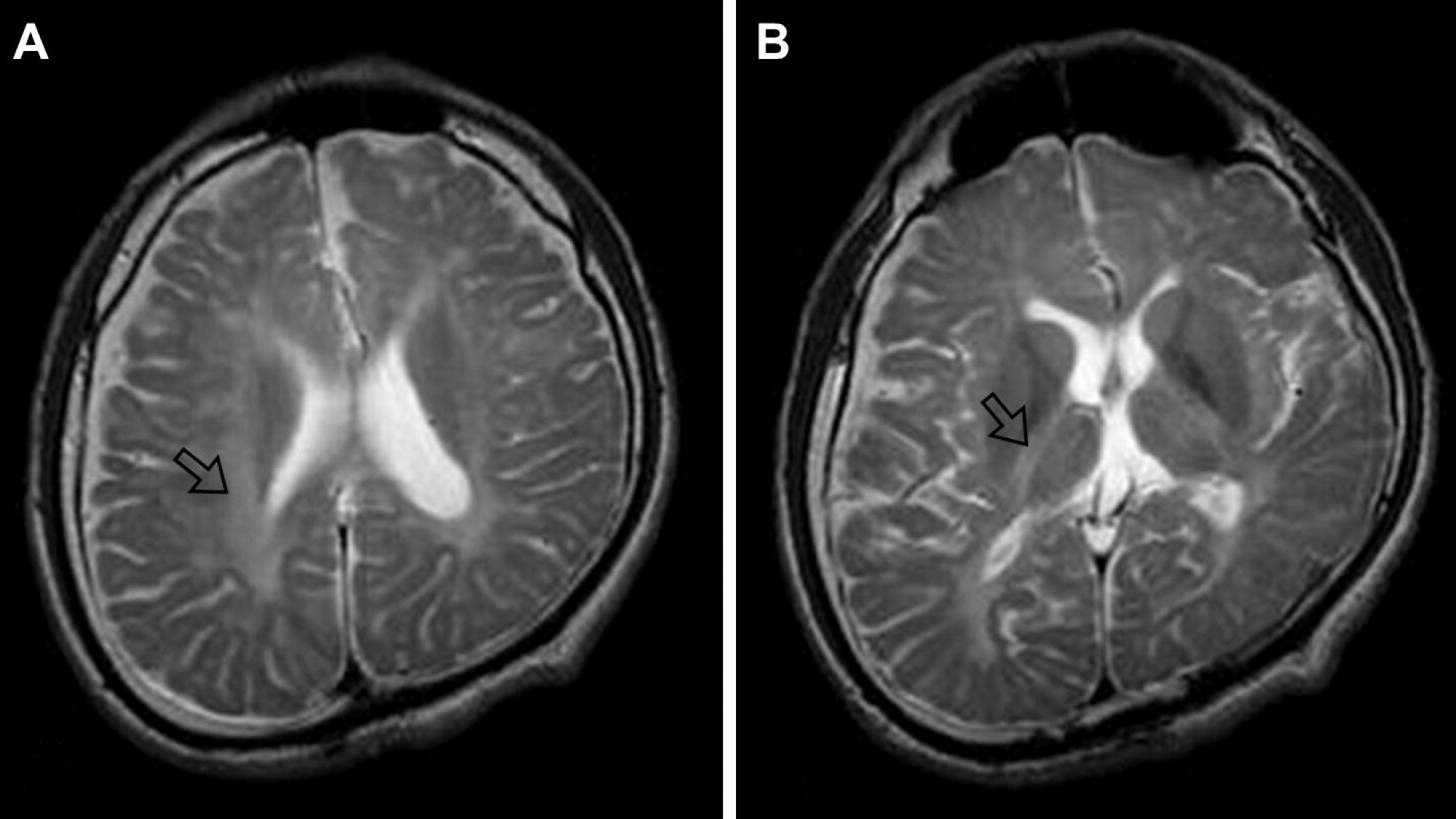


بیماری پلیزائوس-مرزباخر (Pelizaeus-Merzbacher) نوعی اختلال است که بر مغز و نخاع تأثیر می گذارد و با فراوانی 1 به 200 هزار تا 500 هزار نفر در جمعیت آمریکا بروز می کند.
این بیماری نوعی لوکودیستروفی است و با مشکلات مربوط به هماهنگی ، مهارت های حرکتی و یادگیری مشخص می شود. سن شروع و شدت علائم، بسته به نوع بیماری متفاوت است.
این موضوع به دلیل عدم توانایی در ایجاد میلین به علت جهش در ژن PLP1 ایجاد می شود. الگوی توارث این بیماری وابسته به X مغلوب می باشد و به طور غالب در فرزندان پسر بروز می کند.
علائم
بیماری پلیزائوس-مرزباخر به انواع کلاسیک و شدید (کاناتال) تقسیم می شود. اگرچه این دو نوع از نظر شدت متفاوت هستند اما علائم آنها با هم همپوشانی دارند. نوع کلاسیک یک شایع تر است.
درافراد مبتلا به نوع کلاسیک پلیزائوس-مرزباخر، در طول سال اول زندگی احساس ضعف عضلانی (هیپوتونی) و حرکات غیر ارادی چشم ها (نیستاگموس) و تاخیر در شروع مهارت های حرکتی مانند نشستن یا راه رفتن دیده می شود.
با بزرگتر شدن کودک ، ممکن است نیستاگموس بهبود یابد ، اما اختلالات حرکتی دیگری نظیر سفتی عضلات (اسپاسم) ، مشکلات مربوط به تعادل (آتاکسی) و حرکات غیر ارادی ایجاد می شود.
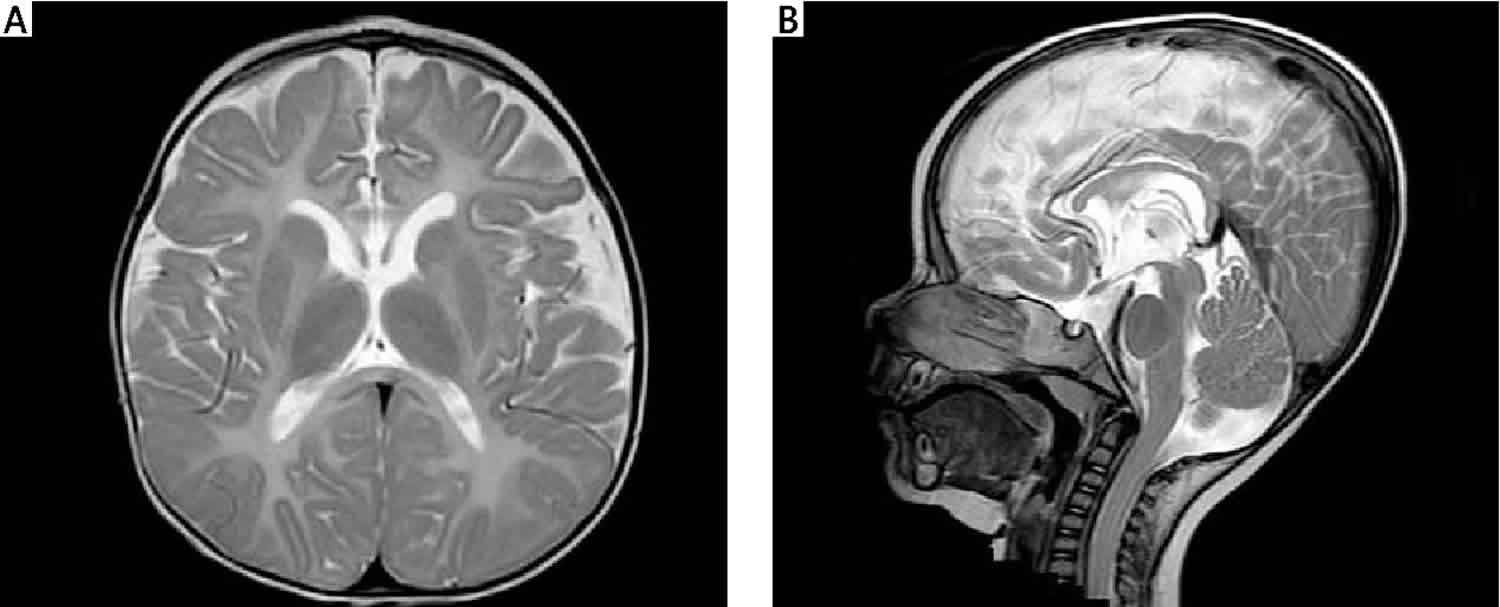
بیماری پلیزائوس-مرزباخر (Pelizaeus-Merzbacher) نوعی اختلال است که بر مغز و نخاع تأثیر می گذارد و با فراوانی 1 به 200هزار تا 500 هزار نفر در جمعیت آمریکا بروز می کند.
این بیماری نوعی لوکودیستروفی است و با مشکلات مربوط به هماهنگی ، مهارت های حرکتی و یادگیری مشخص می شود. سن شروع و شدت علائم، بسته به نوع بیماری متفاوت است.
این موضوع به دلیل عدم توانایی در ایجاد میلین به علت جهش در ژن PLP1 ایجاد می شود. الگوی توارث این بیماری وابسته به X مغلوب می باشد و به طور غالب در فرزندان پسر بروز می کند.
علت بیماری
بیماری پلیزائوس-مرزباخر در اثر جهش در ژن PLP1 ایجاد می شود. این ژن دستورالعمل هایی برای ساخت پروتئین پروتئو لیپید 1 (PLP1) و ایزوفرم این پروتئین ، به نام DM20 را ارائه می دهد.
PLP1 و DM20 اساسا در سیستم عصبی مرکزی بیان می شوند. پروتئین های اصلی موجود در میلین (پوشش عایق رشته های عصبی) هستند.
کمبود این پروتئین می تواند باعث از بین رفتن غلاف میلین گردد ، که به نوبه خود عملکرد سیستم عصبی را مختل می کند . در نتیجه علائم و نشانه های بیماری پلیزائوس-مرزباخر تظاهر می یابد.
تخمین زده می شود که در 5 تا 20 درصد از افراد مبتلا به این بیماری جهش در ژن PLP1 شناسایی نشده است. در این موارد ، علت این بیماری ناشناخته است.
الگوی وراثت
با توجه به اینکه ژن مسئول این بیماری بر روی کروموزوم جنسی X قرار دارد، در نتیجه با الگوی وابسته به جنس مغلوب توارث می یابد.
در مردان (که فقط یک کروموزوم X دارند) ، یک نسخه تغییر یافته از ژن در هر سلول برای ایجاد این بیماری کافی است.
. از آنجا که خانم ها دو نسخه از کروموزوم X دارند ، یک کپی تغییر یافته از ژن در هر سلول معمولاً علائم خفیف تری نسبت به آقایان دارد یا ممکن است هیچ علائمی ایجاد نکند.
تشخیص
تشخیص برای یک بیماری ژنتیکی یا نادر اغلب می تواند چالش برانگیز باشد. متخصصان به منظور تشخیص به طور معمول سابقه پزشکی ، علائم ، معاینه فیزیکی و نتایج آزمایشگاه فرد را بررسی می کنند.
ثبت آزمایش های ژنتیکی (Genetic Testing Registry) اطلاعاتی در مورد آزمایشات ژنتیکی برای این بیماری های نادر از جمه پلیزائوس-مرزباخر فراهم می کند.
مخاطبان مورد نظر برای GTR متخصصان ژنتیک و محققان هستند. بیماران در صورت داشتن سوالات خاص در مورد آزمایش ژنتیک باید با متخصص ژنتیک تماس بگیرند.
درمان
هیچ درمان مشخصی برای بیماری پلیزائوس-مرزباخر وجود ندارد . مدیریت بیماری معمولاً توسط یک تیم پزشکی متشکل از متخصصان مغز و اعصاب ، طب فیزیکی ، ارتوپدی ، پزشکی ریوی و گوارش است.
تاکتیک های مدیریتی ممکن است شامل گاستروستومی (برای افراد مبتلا به دیسفاژی شدید)، داروهای ضد صرع (AED) برای تشنج، و مدیریت عادی اسپاستیسیته از طریق فیزیوتراپی ، ورزش ، داروها (باکلوفن ، دیازپام ، تیزانیدین ، سم بوتولینوم) ، ارتزها (Orthosis) و در موارد شدید جراحی جراحی باشد. افراد مبتلا به اسکولیوز ممکن است از نشستن با ویلچر و فیزیوتراپی بهره مند شوند .
پیش آگهی
پیش آگهی برای مبتلایان به بیماری پلیزائوس-مرزباخر ضعیف است ، و تا قبل از مرگ رو به وخامت است. افراد مبتلا به نوع شدید (مادرزادی) ممکن است در دوران نوزادی یا کودکی بر اثر آسپیراسیون ریوی بمیرند .
. با این حال مراقبت های دقیق ، این افراد ممکن است تا دهه سوم یا بیشتر زندگی کنند. زنده ماندن تا دهه ششم یا هفتم برای افرادی با نوع کلاسیک مشاهده شده است.
روش تشخیص ژنتیکی
برای تشخیص ژنتیک بیماران ابتدا بررسی حذف و مضاعف شدگی در ژن PLP1 توصیه می شود. کارایی این روش در یافتن جهش منجر به بیماری بین 60 تا 70 % می باشد. در صورتیکه بیمار فاقد حذف و اضافه شدگی در این ژن باشد، تعیین توالی کل ژن برای پیدا کردن 30% مابقی جهش های نقطه ای توصیه می گردد.
در مواردی که تشخیص قطعی وجود ندارد، برای صرفه جویی در زمان و هزینه با استفاده از تکنیک تعیین توالی هول اگزوم ژن عامل این بیماری و سایر بیماریهای با علایم مشابه در یک پنل چند ژنی مورد بررسی قرار می گیرد.
از آنجاییکه این ژن بر روی کروموزوم X واقع شده است صرف نظر از ازدواج فامیلی به طور مستقل می تواند از مادر ناقل به احتمال 50 % به فرزندان پسر انتقال یابد و باعث ابتلای آنها به این بیماری گردد.
هیچ یک از دختران این مادر مبتلا به بیماری نمی شوند. اما به احتمال 50% می توانند مشابه مادر، ناقل بیماری مذکور باشند.
[…] http://www.GenomeLab.ir […]